Альдегиды
Альдеги́ды (от лат. alcohol dehydrogenatus — спирт, лишённый водорода) — класс органических соединений, содержащих альдегидную группу (-CHO)[1]. ИЮПАК определяет альдегиды как вещества вида R-CHO, в которых карбонильная группа связана с одним атомом водорода и одной группой R[2].

Названия альдегидов
Этимология
Слово альдегид было придумано Юстусом фон Либихом как сокращение латинского alcohol dehydrogenatus — дегидрированный спирт[3] (в некоторых источниках — alcohol dehydrogenatum[1]). Название радикала формил, а также другие однокоренные слова (формальдегид, формиаты), произошли от лат. formica — муравей[4].
Тривиальные названия
В популярной и научной литературе можно нередко встретить исторические, или тривиальные, названия альдегидов, которые вследствие сложившейся традиции используются вместо систематических названий. Тривиальные названия обычно происходят от названия соответствующих карбоновых кислот, а также от названия источника, из которого был выделен тот или иной альдегид. Так, например, формальдегид называют муравьиным альдегидом, этаналь — уксусным, пентаналь — валериановым альдегидом, цитронеллаль получил своё название, поскольку был выделен из масла цитрусовых.
Исторически сложилось, что парфюмеры называют многие пахучие вещества альдегидами, даже те, которые не имеют ничего общего с ними. Среди таковых, например, персиковый, земляничный и кокосовый альдегид, которые являются не альдегидами, а сложными эфирами или лактонами. Также некоторые альдегиды традиционно называются по числу атомов углерода, например, персиковый альдегид, обозначаемый как «альдегид C14», на самом деле имеет лишь 11 атомов углерода[5].
Систематическая номенклатура
По номенклатуре ИЮПАК названия простых альдегидов образуются от названий соответствующих алканов с добавлением суффикса -аль, а диальдегидов — суффикса -диаль (в данном случае атом углерода альдегидной группы уже входит в состав родоначального алкана). При этом в названии номер при альдегидной группе, как правило, не ставят, поскольку она всегда занимает крайнее положение. Если карбонильная группа не входит в родоначальную структуру (например, если родоначальной структурой является циклический углеводород или гетероцикл), то к названию добавляется суффикс -карбальдегид[6][7].
Если в данном соединении альдегидная группа не является старшей, то в таких случаях её обозначают используя приставку формил-, указывая её положение[7].
В устаревших Женевской (1892) и Льежской (1930) номенклатурах, впоследствии заменённых систематической номенклатурой ИЮПАК, альдегиды обозначались при помощи суффикса -ал[8].

Классификация альдегидов
Альдегиды классифицируются следующим образом (в скобках приведены примеры)[9]:
- В зависимости от насыщенности углеводородного заместителя:
- предельные (насыщенные) альдегиды (ацетальдегид);
- непредельные (ненасыщенные) альдегиды (акролеин);
- ароматические альдегиды (бензальдегид).
- По числу карбонильных групп:
- альдегиды с одной карбонильной группой (формальдегид);
- диальдегиды (глиоксаль);
- многоатомные альдегиды.
Нахождение в природе

Альдегидная группа содержится во многих природных веществах, таких, как углеводы (альдозы), некоторые витамины (ретиналь, пиридоксаль). Их следы содержатся в эфирных маслах и часто способствуют их приятному запаху, например, коричный альдегид (в кассиевом масле его может быть до 75 %, а в цейлонском коричном масле даже до 90 %) и ванилин.
Алифатический альдегид СН3(СН2)7С(Н)=О (тривиальное название — пеларгоновый альдегид) содержится в эфирных маслах цитрусовых растений, обладает запахом апельсина, его используют как пищевой ароматизатор[10].
Цитраль содержится в лемонграссовом и кориандровом маслах (до 80 %), цитронеллаль — в цитронелловом (приблизительно 30 %) и эвкалиптовом, бензальдегид — в масле горького миндаля. Куминовый альдегид содержится в масле тмина, гелиотропин — в масле гелитропа и сирени, анисовый альдегид и жасминальдегид в небольших количествах содержатся во многих эфирных маслах[1][5].
Методы синтеза альдегидов
Окислительные методы
- Для получения альдегидов в лабораторных условиях часто используется реакция окисления первичных спиртов реагентами, представляющими собой комплексные соединения оксида хрома(VI) с третичными аминами, в частности, лучшими реагентами являются комплекс с пиридином (CrO3 · 2C5H5N, реагент Саррета — Коллинза) и хлорхромат пиридиния (C5H5NH+CrO3Cl-, реагент Кори, PCC). Данные реагенты позволяют получать альдегиды с высоким выходом, а хлорхромат пиридиния также не затрагивает двойную связь. Для этих же целей применяют и другие селективные окислители, например оксид марганца(IV) MnO2, карбонат серебра на цеолите, а также диметилсульфоксид в присутствии основания (окисление по Сверну)[11].
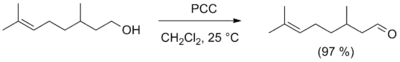
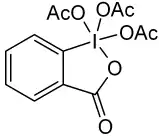
- Реакция окисления периодинаном Десса-Мартина. Первичные спирты при этом селективно окисляются до альдегидов.[12]

- Как метод получения альдегидов может использоваться восстановительный озонолиз симметричных дизамещённых алкенов либо циклических алкенов (в данном случае реакция приводит к образованию диальдегида). Аналогичное превращение может быть проведено под действием смеси OsO4 и NaIO4[13].
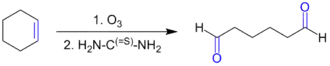
- Также к данному типу реакций относится окисление вицинальных диолов йодной кислотой или тетраацетатом свинца[14].

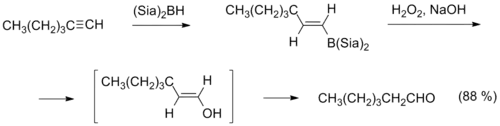
- Важным методом также является гидроборирование — окисление алкинов, в ходе которого к алкину против правила Марковникова присоединяется диалкилборан (например, дисиамилборан), а полученный продукт окисляется щелочным раствором пероксида водорода, что приводит к образованию альдегида[15].
Восстановительные методы
Ряд производных карбоновых кислот (хлорангидриды, сложные эфиры, нитрилы, амиды) могут быть восстановлены до альдегидов под действием специфических восстановителей[16].
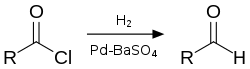
- Так, например, в реакции Розенмунда хлорангидриды восстанавливают под действием водорода на палладиевом катализаторе. Аналогичное превращение можно провести под действием три(трет-бутокси)алюмогидрида лития[17].
- Сложные эфиры селективно восстанавливаются до альдегидов под действием диизобутилалюминийгидрида[18].
Синтез ароматических альдегидов
Ароматические альдегиды могут быть синтезированы принципиально отличными методами, основанными на реакциях ароматического электрофильного замещения.
- Альдегидную группу можно ввести в ароматические соединения реакциями Гаттермана, Гаттермана — Коха, Вильсмейера — Хаака, Рихе и Раймера — Тимана. Исторически первая реакция Гаттермана — Коха (1897) применима к бензолу и его алкилзамещённым производным, которые вступают в реакцию с оксидом углерода(II) CO и хлороводородом HCl в присутствии AlCl3 и CuCl, давая соответствующие бензальдегиды (альдегидная группа вводится в пара-положение). Улучшенный метод (реакция Гаттермана) состоит в использовании цианида цинка Zn(CN)2 и соляной кислоты и позволяет формилировать фенолы и гетероароматические соединения. Для формилирования фенолов также используется реакция Раймера — Тимана. Введение альдегидной группы в ароматические ядра, активированные гидроксильной, алкоксильной или диалкиламинной группой, осуществляется по реакции Вильсмейера — Хаака с использованием диметилформамида и хлорокиси фосфора (или аналогичных реагентов)[19].

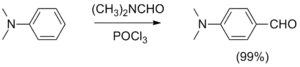
- Также ароматические альдегиды могут быть получены окислением метилзамещённых бензолов под действием ряда окислителей, в том числе оксида хрома(VI) CrO3, оксида марганца(IV) MnO2 и нитрата церия — аммония[20].
- Реакция Соммле позволяет окислять бензилгалогениды ArCH2X под действием уротропина с последующим гидролизом образующейся соли до альдегида. Данная реакция применима для синтеза разнообразных ароматических и гетероциклических альдегидов. Подобное превращение можно осуществить, также окисляя бензилгалогениды солями 2-нитропропана[20].

- Ароматические альдегиды можно получать из производных ароматических карбоновых кислот общими методами, однако существуют и специфические реакции. Например, реакция Стефена позволяет восстанавливать ароматические нитрилы хлоридом олова(II) SnCl2 с последующим гидролизом, что приводит к ароматическому альдегиду[20].
Другие методы
Альдегиды также можно получать реакциями гидратации алкинов (реакция Кучерова), пиролизом карбоновых кислот и их смесей в виде паров над оксидами некоторых металлов (ThO2, MnO2, CaO, ZnO) при 400—500 °C, гидролизом геминальных дигалогенопроизводных (если атомы галогена находятся у одного из крайних атомов углерода) и другими реакциями[9].
Промышленные методы получения альдегидов
Известно много методов синтеза альдегидов, однако их использование в промышленности зависит во многом от доступности исходного сырья. Основными промышленными методами получения насыщенных алифатических альдегидов являются[21]:
- гидроформилирование алкенов (оксосинтез);
- дегидрирование или окисление первичных спиртов;
- гидратация ацетилена;
- окисление этилена;
- окисление насыщенных углеводородов (С3, С4).
Также большое значение имеют некоторые специфические синтезы альдегидов, широко применяемых в парфюмерной промышленности[21].
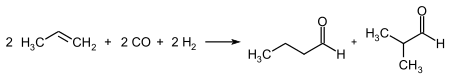
Оксосинтез является наиболее важным процессом для получения альдегидов с тремя атомами углерода и выше. В этой реакции алкены реагируют с синтез-газом (CO + H2) с образованием альдегида, содержащего на один атом углерода больше, чем исходный алкен. При использовании несимметричных алкенов образуется смесь продуктов, соотношение которых можно варьировать путём подбора катализатора[21].
Среди процессов отщепления водорода от первичных спиртов различают дегидрирование, окисление и окислительное дегидрирование. Дегидрирование спиртов проводят при атмосферном давлении и температуре 250—400 °С в присутствии медного или серебряного катализатора. В ходе процесса выделяется водород, который можно использовать без очистки в других процессах. Дегидрирование имеет коммерческое значение в получении уксусного альдегида из этанола: реакцию проводят при 270—300 °С на медном катализаторе, активированном церием, при этом за цикл превращается 25—50 % этанола с селективностью 90—95 %. Побочными продуктами являются этилацетат, этилен, кротоновый альдегид и высшие спирты. Окисление спиртов проводится в избытке воздуха или кислорода при 350—450 °С на катализаторе, содержащем оксиды железа и молибдена. Процесс используется в производстве формальдегида. Данные процессы также применяются в синтезе душистых альдегидов[21].
Окисление алкенов является основным промышленным методом получения ацетальдегида и акролеина. В первом случае окислению подвергается этилен в присутствии хлоридов палладия и меди Вакер-процесс [21][9].



Процесс получения ацетальдегида, основанный на гидратации ацетилена, в последнее время потерял былое значение. Последние фабрики в Западной Европе, синтезирующие ацетальдегид по данной схеме, были закрыты в 1980 году. Причиной этому послужила бо́льшая доступность этилена в качестве сырья, а также токсичность катализатора — сульфата ртути[21].
Ежегодное мировое производство формальдегида (по данным на 1996 год) составило 8,7·106 т[22], ацетальдегида (на 2003 год) — 1,3·106 т[23].
Основным методом получения бензальдегида является гидролиз бензальхлорида в кислой или щелочной средах. В качестве гидролизующих агентов могут применяться гидроксид кальция, карбонат кальция, гидрокарбонат натрия, карбонат натрия, а также различные кислоты с добавлением солей металлов. Исходное сырьё, в свою очередь, получают при хлорировании толуола в боковую цепь. Менее распространённый процесс основан на частичном окислении толуола[24].
Физические свойства альдегидов
Формальдегид представляет собой газообразное при комнатной температуре вещество. Альдегиды до С12 — жидкости, а альдегиды нормального строения с более длинным неразветвлённым углеродным скелетом являются твёрдыми веществами.
Температуры кипения альдегидов с неразветвлённым строением углеродной цепи выше, чем у их изомеров. Например, валериановый альдегид кипит при 100,4 °C, а изовалериановый — при 92,5 °C. Они кипят при более низких температурах, чем спирты с тем же числом углеродных атомов, например, пропионовый альдегид кипит при 48,8 °C, а пропанол-1 при 97,8 °C. Это показывает, что альдегиды, в отличие от спиртов, не являются сильно ассоциированными жидкостями[9]. Данное свойство используется в синтезе альдегидов путём восстановления спиртов: поскольку температура кипения альдегидов в целом ниже, они могут быть легко отделены и очищены от спирта перегонкой[25]. В то же время их температуры кипения намного выше, чем у углеводородов с той же молекулярной массой, что связано с их высокой полярностью[9].
Вязкость, плотность и показатель преломления при 20 °C увеличиваются с увеличением молярной массы альдегидов. Низшие альдегиды являются подвижными жидкостями, а альдегиды от гептаналя до ундеканаля имеют маслообразную консистенцию[25].
Формальдегид и ацетальдегид практически неограниченно смешиваются с водой, однако, с ростом длины углеродного скелета, растворимость альдегидов в воде сильно уменьшается, например, растворимость гексаналя при 20 °С составляет лишь 0,6 % по массе. Алифатические альдегиды растворимы в спиртах, простых эфирах и других распространённых органических растворителях[25].
Низшие альдегиды имеют резкий запах, а высшие гомологи (С8-С13) являются компонентами многих парфюмерных изделий[25].
| Название | Формула | Температура плавления, °C | Температура кипения, °C | Плотность, г/см³ (при 20 °C) |
|---|---|---|---|---|
| Формальдегид | HCHO | −93 | −21 | 0,82 (при –20 °С) |
| Ацетальдегид | CH3CHO | −123 | 21 | 0,778 |
| Пропаналь | CH3CH2CHO | −81 | 49 | 0,797 |
| Бутаналь | CH3CH2CH2CHO | −99 | 76 | 0,803 |
| Акролеин | CH2=CH–CHO | −88 | 53 | 0,841 |
| Кротоновый альдегид | CH3-CH=CH–CHO | −74 | 104 | 0,852 |
| Бензальдегид | C6H5CHO | −56 | 179 | 1,05 |
| Салициловый альдегид | о-HO–C6H4CHO | 2 | 197 | 1,16 |
| Ванилин |
| 82 | 285 | — |
Строение
Атом углерода в карбонильной группе находится в состоянии sp2-гибридизации. Углы R-C-H, R-C-O и H-C-O составляют приблизительно 120° (где R — алкил).
Двойная связь карбонильной группы сходна по физической природе с двойной связью между углеродными атомами, однако в то же время энергия связи С=О (749,4 кДж/моль) больше, чем энергия двух простых связей (2×358 кДж/моль) C-O. С другой стороны, кислород является более электроотрицательным элементом, чем углерод, и потому электронная плотность вблизи атома кислорода больше, чем вблизи атома углерода. Дипольный момент карбонильной группы составляет ~9⋅10−30 Кл·м[9]. Длина связи С=О составляет 0,122 нм[14].
Поляризация двойной связи «углерод-кислород» по принципу мезомерного сопряжения позволяет записать следующие резонансные структуры:

Подобное разделение зарядов подтверждается физическими методами исследования и во многом определяет реакционную способность альдегидов как выраженных электрофилов и позволяет им вступать в многочисленные реакции нуклеофильного присоединения[28].
Химические свойства
Высокая реакционная способность связана с наличием полярной связи С=О. Альдегиды являются жёсткими основаниями Льюиса и, в соответствии с этим, атом кислорода в них может координироваться с жёсткими кислотами: H+, ZnCl2, BF3, AlCl3 и т. д.[14] В общем случае химические свойства альдегидов аналогичны кетонам, однако альдегиды проявляют бо́льшую активность, что связано с большей поляризацией связи. Кроме того, для альдегидов характерны реакции, не характерные для кетонов, например гидратация в водном растворе.
Реакции присоединения к карбонильной группе
Альдегиды содержат поляризованную карбонильную группу и склонны присоединять нуклеофильные реагенты, как нейтральные (аммиак, амины, воду, спирты, тиолы и др.), так и анионные (цианид-ион CN-, алкоголяты, гидрид-ион H-, карбанионы и др.). За исключением реакций восстановления гидридами типа алюмогидрида лития LiAlH4, а также взаимодействия с реактивами Гриньяра, данные процессы являются обратимыми. Необходимо также различать два типа обратимых реакций присоединения: первый тип приводит к образованию тетраэдрического продукта присоединения, а второй тип включает в себя также последующую реакцию дегидратации, в результате которой происходит образование двойной связи между электрофильным атомом углерода и нуклеофилом. Реакции второго типа характерны, в основном, для азотсодержащих нуклеофилов[29].

В данных реакциях альдегиды являются более реакционноспособными по сравнению с кетонами. Это связано с большей термодинамической устойчивостью кетонов, а также меньшими пространственными затруднениями в случае присоединения к альдегидам[29].
Простейшей модельной реакцией данного типа является реакция гидратации альдегидов, протекающая в их водных растворах. Согласно правилу Эльтекова — Эрленмейера, образующиеся при этом 1,1-диолы неустойчивы и с отщеплением молекулы воды превращаются обратно в исходные карбонильные соединения. Гидратация наблюдается в существенной степени лишь для низших альдегидов. Так, формальдегид гидратирован на 99,999 %, ацетальдегид — на 58 %. Когда положительный заряд на атоме углерода увеличивается в достаточной степени за счёт связанных с ним радикалов, 1,1-диолы становятся устойчивыми и могут быть выделены (например, хлораль легко присоединяет воду с образованием устойчивого аддукта — хлоральгидрата). Реакция гидратации катализируется как кислотами, так и основаниями[14][30].

Подобным образом протекает и реакция присоединения спиртов по карбонильной группе, имеющая важное значение в органическом синтезе для защиты карбонильной группы. Первичный продукт присоединения называется полуацеталем, далее под действием кислоты он превращается в ацеталь. При стоянии альдегиды также образуют циклические или полимерные ацетали (например, триоксан или параформ для формальдегида и паральдегид для ацетальдегида). При нагревании этих соединений со следовыми количествами кислот происходит деполимеризация и регенерация исходных альдегидов[31].

Аналогичные превращения происходят также с участием серосодержащих аналогов спиртов — тиолов; они приводят, соответственно, к тиоацеталям, также играющим важную роль в тонком органическом синтезе[31].
Альдегиды могут присоединять циановодород HCN с образованием циангидринов, применяемых в органическом синтезе для получения α,β-ненасыщенных соединений, α-гидроксикислот, α-аминокислот. Данная реакция также является обратимой и катализируется основаниями. В лабораторных условиях циановодород (т. кип. 26 °C) обычно получают действием эквивалентного количества минеральной кислоты на цианид натрия или калия[32].
Относительно небольшие пространственные затруднения при присоединении нуклеофилов к альдегидам позволяют превращать их в бисульфитные производные под действием большого избытка гидросульфита натрия NaHSO3. Данные соединения представляют собой кристаллические вещества и часто используются для выделения, очистки или хранения соответствующих альдегидов, поскольку последние могут быть легко из них регенерированы под действием кислоты или основания[32].

Реакция альдегидов с магний- и литийорганическими соединениями приводит к образованию вторичных спиртов (в случае формальдегида — первичных). Процесс может осложняться побочными реакциями енолизации и восстановления карбонильного соединения, которые приводят к снижению выхода. При использовании литийорганических соединений эти помехи удаётся устранить[33].
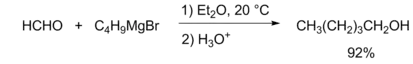

При реакции альдегидов с первичными и вторичными аминами происходит образование иминов и енаминов соответственно. В основе обеих реакций лежит присоединение нуклеофильных реагентов по карбонильной группе с последующим отщеплением воды от полученного тетраэдрического интермедиата. Реакция образования иминов требует кислотного катализа и наиболее эффективно протекает в области pH от 3 до 5. Для получения енаминов с удовлетворительным выходом необходимо применять азеотропную отгонку воды, что позволяет сместить равновесие в сторону образования продукта. Обычно в качестве вторичных аминов используют циклические амины (пирролидин, пиперидин или морфолин)[34].

Аналогичным образом альдегиды реагируют с гидроксиламином, гидразином, 2,4-динитрофенилгидразином, семикарбазидом и другими подобными соединениями. Большинство получаемых при этом соединений являются кристаллическими и могут быть использованы для идентификации альдегидов по температуре плавления и другим характеристикам. Также эти соединения находят применение в органическом синтезе, например, гидразоны могут быть восстановлены по реакции Кижнера — Вольфа[34].
Реакции сопряжённого присоединения
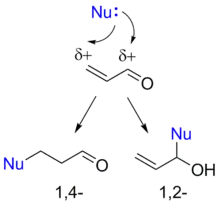
Присоединение нуклеофильных реагентов к α,β-ненасыщенным альдегидам может протекать как по карбонильной группе, так и по «четвёртому» положению сопряжённой системы. Причина этого заключается в том, что двойная углерод-углеродная связь поляризуется под действием полярной карбонильной группы (мезомерный эффект), и дальний от карбонильной группы атом углерода двойной связи приобретает частичный положительный заряд. Реакция нуклеофила с данным атомом углерода называется сопряжённым присоединением, или 1,4-присоединением. Присоединение к карбонильной группе по аналогии называют 1,2-присоединением. Формальным результатом 1,4-присоединения является присоединение нуклеофила по углерод-углеродной двойной связи. Во многих случаях 1,2- и 1,4-присоединение являются конкурирующими реакциями, однако иногда удаётся проводить селективные реакции с получением продуктов 1,2- либо 1,4-присоединения[35].
Присоединение первичных и вторичных аминов к α,β-ненасыщенным альдегидам протекает в мягких условиях и приводит к образованию 1,4-продукта. Напротив, в случае циановодорода наблюдается конкурентное образование обоих продуктов с преобладанием продукта 1,2-присоединения. Чтобы в данной реакции исключить возможность 1,2-присоединения, используют специальный реагент — диэтилалюминийцианид (C2H5)2AlCN[36].
Литийорганические соединения присоединяются исключительно по карбонильной группе, давая аллиловые спирты. Сопряжённое присоединение проводят под действием медьорганических реагентов — диалкилкупратов, которые позволяют ввести в карбонильное соединение не только первичную, но также вторичную или третичную алкильную, алкенильную или арильную группу. Магнийорганические реагенты (реактивы Гриньяра), полученные из магния сверхвысокой чистоты, также присоединяются с образованием 1,2-продуктов, в то время как обыкновенные реактивы Гриньяра, предположительно из-за примесей других металлов (например, меди и железа) вступают и в 1,2-, и в 1,4-присоединение, что регулируется пространственными факторами. В настоящее время магнийорганические реагенты утратили своё значение в данной области[37].
Борорганические соединения (триалкилбораны) реагируют с непредельными альдегидами, давая продукты 1,4-присоединения[38]
Реакции α-метиленового звена
Альдегиды вступают в реакцию с галогенами (хлором, бромом или иодом), образуя галогенпроизводные, при этом галогенирование осуществляется исключительно в α-положение (в положение, соседнее с карбонильной группой)[39].
Альдегиды проявляют свойства слабых кислот: под действием оснований они способны отщеплять протон от α-метиленовой группы, превращаясь в енолят-ион. Обычно для достаточно полного депротонирования используют сильные основания (гидрид натрия, гидрид калия, диизопропиламид лития и др.) в апротонных растворителях (тетрагидрофуран, ДМСО). Превращение карбонильной формы альдегидов в енольную форму протекает и в отсутствие сильных оснований. Образующиеся при этом енолы, как правило, намного менее стабильны, чем карбонильная форма, например, для ацетальдегида константа равновесия — только 6⋅10−5 при комнатной температуре[40]). Данное равновесие, существующее между карбонильной и енольной формами называется кето-енольной таутомерией[41].
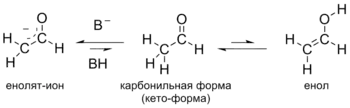
Благодаря способности образовывать енолят-ионы альдегиды вступают в ряд химических реакций, где эти частицы выступают как нуклеофилы. В частности, для них характерны реакции конденсации. В слабоосновной среде (в присутствии ацетата, карбоната или сульфита калия) подвергаются альдольной конденсации, в ходе которой часть молекул альдегида выступает как карбонильная компонента (реагирует карбонильной группой), а часть молекул альдегида под действием основания превращается в енолят-ионы и выступает как метиленовая компонента (вступает в реакцию α-метиленовым звеном). Образующийся альдоль при нагревании отщепляет воду с образованием α,β-непредельного альдегида (переход от предельного альдегида к непредельному через альдоль называется кротоновой конденсацией или альдольно-кротоновой конденсацией)[9][42].
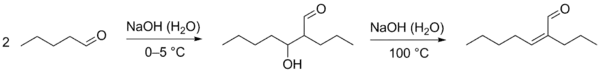
При реакции между двумя разными альдегидами образуется смесь четырёх различных альдолей. Исключение составляют случаи, когда разделение реагентов на карбонильную и метиленовую компоненту очевидно (например, один из альдегидов не содержит α-метиленового звена и может выполнять роль только карбонильной компоненты). Разработаны также методы повышения селективности подобных реакций. Перекрёстная конденсация ароматических альдегидов с кетонами, получила название реакции Кляйзена — Шмидта[42]. Известны также схожие реакции альдегидов: реакция Кнёвенагеля, реакция Тищенко, реакция Перкина, бензоиновая конденсация и другие[1].
Реакции окисления
Окисление альдегидов до соответствующих карбоновых кислот кислородом протекает по радикально-цепному механизму (автоокисление) с образованием промежуточных продуктов — пероксокислот.
Альдегиды легко окисляются до соответствующих карбоновых кислот под действием разнообразных окислителей. Наиболее часто используются перманганат калия, а также реагент Джонса (CrO3 + H2SO4), который даёт наилучшие результаты (в течение короткого времени при низкой температуре достигается более чем 80%-ый выход карбоновой кислоты). Реагент Джонса также не лишён недостатков, в частности, он недостаточно селективен и окисляет другие функциональные группы, а кислая среда способствует нежелательной изомеризации или разложению субстрата[43].

Избежать этих проблем можно при использовании более мягкого окислителя — реактива Толленса (аммиачного раствора оксида серебра), который не затрагивает кратные связи и гидроксильные группы спиртов. Эта реакция широко применяется для обнаружения альдегидов (реакция «серебряного зеркала»)[43].
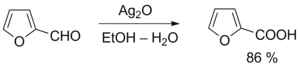
Окисление метиленовых групп в α-положении альдегидов диоксидом селена приводит к образованию 1,2-дикарбонильных соединений[14][43].
Альдегиды медленно окисляются на воздухе при комнатной температуре. Этот радикальный процесс ускоряется при облучении или в присутствии ионов Fe2+. Ароматические альдегиды подвергаются окислению легче, чем алифатические. Данная реакция не имеет синтетического значения, однако её протекание необходимо учитывать при хранении альдегидов: желательно хранить их в темноте и инертной атмосфере[44].
Ароматические альдегиды также окисляются до карбоновых кислот или сложных эфиров фенолов (реакция Байера — Виллигера) под действием надкислот, причём соотношение продуктов зависит как от заместителей в ароматическом ядре, так и от кислотности среды[44].
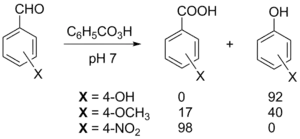
Реакции восстановления
Альдегиды можно восстанавливать до первичных спиртов. Наиболее распространённые методы восстановления включают реакции с комплексными гидридами: боргидридом натрия NaBH4, боргидридом лития LiBH4 и алюмогидридом лития LiAlH4. Боргидрид натрия является более избирательным реагентом и позволяет восстанавливать карбонильную группу альдегидов и кетонов, не затрагивая сложноэфирные, нитрильные, амидные, лактонные и оксирановые группы. Он также не восстанавливает изолированную двойную углерод-углеродную связь. Алюмогидрид лития менее селективен и восстанавливает перечисленные выше функциональные группы, поэтому восстановление альдегидов с его применением возможно только в отсутствие этих групп[45].
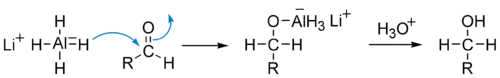
Историческую роль играет реакция Меервейна — Пондорфа — Верлея, в которой в качестве восстановителя используется изопропилат алюминия. В настоящее время этот метод вытеснен более эффективной реакцией восстановления альдегидов и кетонов изопропиловым спиртом в присутствии окиси алюминия[45].
Алифатические альдегиды обычно не гидрируют на палладиевых катализаторах, но для этих целей можно использовать рутений на угле, никель Ренея или платину[45].
Альдегиды как основания Льюиса
В соответствии с наличием неподелённых электронных пар атома кислорода карбонильной группы альдегиды являются жёсткими основаниями Льюиса и, в соответствии с этим, атом кислорода в них может координироваться с жёсткими кислотами: H+, ZnCl2, BF3, AlCl3 и т. д.[14]. В кислой среде альдегиды протонируются с образованием оксониевого катиона. Альдегиды являются очень слабыми основаниями, намного более слабыми, чем вода и спирты, но тем не менее эти свойства играют исключительно важную роль в химических реакциях[46].
Другие реакции
Альдегиды, не имеющие атомов водорода при α-углеродном атоме (то есть имеющие общую формулу R3CCHO) под действием водно-спиртового раствора щёлочи вступают в реакцию Канниццаро, в которой одновременно выступают в роли окислителя и восстановителя. Сфера применения данного процесса расширяется за счёт перекрёстной реакции Канниццаро, протекающей между ароматическим альдегидом и формальдегидом в присутствии щёлочи. При этом восстановителем является формальдегид, а другой альдегид восстанавливается до соответствующего спирта[47].
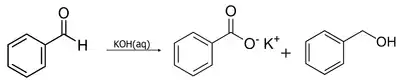
Альдегиды могут вступать в реакции с илидами фосфора по реакции Виттига, образовывая алкены с заданной конфигурацией двойной связи (как правило, образуются Z-алкены, но разработаны и модификации, позволяющие получать E-алкены). В данное время это один из лучших методов региоспецифического синтеза алкенов[48].

Также альдегиды взаимодействуют с илидами серы, давая оксираны (реакция Кори — Чайковского)[49].
Альдегиды подвергаются реакции декарбонилирования в присутствии некоторых комплексов переходных металлов, например, катализатора Уилкинсона[50].
Химические методы идентификации альдегидов
Качественный анализ карбонильных групп
- Тест Бреди — 2,4-динитрофенилгидразин с альдегидами даёт жёлтый, оранжевый (если альдегид алифатический) или красный (если альдегид ароматический) осадок:
- RCHO + C6H3(NO2)2NHNH2 → C6H3(NO2)2NHNCHR + H2O
- Реакция «серебряного зеркала» и реакция с фелинговой жидкостью предназначена для распознавания альдегидов и кетонов — альдегиды окисляются до карбоновых кислот, кетоны с этими реагентами не реагируют.
- Реакция с гидроксидом меди (II), происходящая при нагревании, при этом альдегиды окисляются до карбоновых кислот, а гидроксид меди (II) восстанавливается до оксида меди (I)[51][52]:
- RCHO + 2Cu(OH)2 → RCOOH + Cu2O + 2H2O
- Реактив Шиффа (фуксинсернистая кислота) реагирует с альдегидами с образованием яркоокрашенного фиолетового продукта[53]:

Количественный анализ альдегидов
- При действии хлорида гидроксиламиния образуется соответствующий альдоксим и выделяется эквивалентное количество хлоридной кислоты. Выделившуюся после реакции кислоту титруют щелочью; индикатор — раствор бромфенолового синего (окраска раствора изменяется от желто-зеленой до фиолетово-синей)[54]:

- В аналитической практике используется окисление альдегидов и кетонов йодом в щелочной среде. Йод добавляют в избытке, а затем избыток его оттитровывают тиосульфатом натрия[54]:


Спектральные методы анализа альдегидов
ИК-спектроскопические методы анализа альдегидов
Альдегиды легко идентифицировать по ИК-спектру — он содержит специфические полосы поглощения, относящиеся к валентным колебаниям связи C-H в альдегидной группе: два острых пика, расположенные далеко за пределами области поглощения, характерной для связей C-H обычного типа. Кроме того, в ИК-спектрах альдегидов обычно присутствуют полосы поглощения, обусловленные валентными колебаниями связей С=O и C-H: νС=O=1725-1685 см−1, νС-H=2850; 2750 см−1[14].
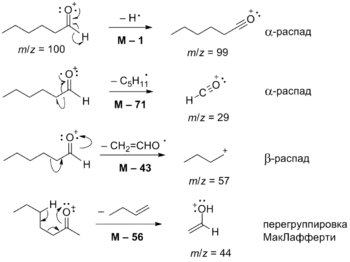
Масс-спектрометрические методы анализа альдегидов
Масс-спектры альдегидов имеют довольно выраженный молекулярный ион, хотя его содержание может быть довольно низким. Потеря алкильных радикалов приводит к образованию ацил-катионов. Для них особенно характерны α- и β-расщепление и перегруппировка Мак-Лафферти[55]. Для альдегидов с подвижным γ-атомом H и не содержащих заместителя у α-углерода характерен пик m/z=44, а для содержащих заместитель появляется интенсивный пик замещённого иона с m/z=44+12n[1][56].
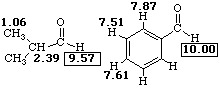
ЯМР-спектроскопические методы анализа альдегидов
В 1Н ЯМР-спектре альдегида наиболее характеристичным является сигнал формильного протона, обычно расположенный в наиболее слабом поле в области δ 9,4—10,1 м д. (9,4-9,7 -алифатические, 9,6-10,1 -ароматические)[1]. Сигнал альдегидной группы в 13C ЯМР-спектре расположен в области 182—215 м д.[57].
УФ-спектроскопические методы анализа альдегидов
Два максимума поглощения от р до р* (<200 нм) и от n до р* (> 200 нм)[57].
Электронно-спектроскопические методы анализа альдегидов
Электронные спектры содержат полосы с λмакс 290 нм для RCHO (R=CH3, C2H5, C3H7), 345 нм для акролеина и 327 для кротонового альдегида[1].
Биологическое действие
Токсичны. Способны накапливаться в организме. Кроме общетоксического, обладают раздражающим и нейротоксическим действием. Эффект зависит от молекулярной массы: чем она больше, тем слабее раздражающее, но сильнее наркотическое действие, причём ненасыщенные альдегиды токсичнее насыщенных. Некоторые обладают канцерогенными свойствами[58].
Альдегиды раздражают слизистые оболочки глаз и верхних дыхательных путей, вредно влияют на нервную систему. С увеличением числа атомов углерода в молекуле раздражающее действие ослабевает. Ненасыщенные альдегиды обладают более сильным раздражающим действием, чем насыщенные.
Ацетальдегид СН3СНО вызывает возбуждение, сменяющееся наркозом. Он является промежуточным продуктом метаболизма этилового спирта в организме. Действие тримера этого альдегида — паральдегида (С2Н4O)3 — сильнее и продолжительнее, в то время как тетрамер — метальдегид (С2Н4O)4 — является более токсичным. Удлинение алкильного радикала в молекуле альдегида приводит к усилению физиологической активности, но вместе с этим возрастает и токсичность[59].
Введение галогена в молекулу альдегида повышает его наркотическое (снотворное) действие. Так, наркотические свойства хлораля более выражены, чем у ацетальдегида. Альдегидная группа усиливает токсичность вещества, но она может быть значительно снижена путём образования гидратной формы альдегида. Гидратные формы мало токсичны, в такой форме хлораль применяется в медицине под названием хлоралгидрата, проявляющего снотворное действие. Введение гидроксильных групп в молекулу альдегида или конденсация их с образованием альдолей существенно снижает реакционную способность, а также физиологическую активность соединений. Так, сахара представляют собой фармакологически инертные вещества. Большинство ароматических альдегидов имеет низкую токсичность, так как они легко окисляются до соответствующих кислот, которые обычно довольно инертны[59].
Лекарственные препараты, содержащие в молекуле альдегидную группу, и их основное действие на организм
| Название | Действие на организм |
|---|---|
| Формальдегид (формалин) | Антисептическое |
| Хлоральгидрат | Снотворное, противосудорожное |
| Цитраль | Снижает артериальное давление |
| Циминаль | Противомикробное |
Применение
Из всех альдегидов больше всего производится формальдегида (около 6 млн тонн/год). Он, в основном, используется в производстве смол — бакелита, галалита (в сочетании с мочевиной, меламином и фенолом), для дубления кож, протравливания зерна. Также из него синтезируют лекарственные средства (уротропин) используют как консервант биологических препаратов (благодаря способности свертывать белок). Он является предшественником метилендифенилдиизоцианата, использующегося в производстве полиуретанов и гексогена (довольно сильной взрывчатки).
Второй по масштабам производства альдегид — масляный альдегид (получают около 2,5 млн тонн/год методом гидроформилирования). Некоторые альдегиды синтезируют только в небольших масштабах (менее 1000 тонн / год) и используют в качестве ингредиентов в парфюмерии и ароматов (в основном альдегиды с числом атомов углерода от 8 до 12)[1]. Например, это коричный альдегид и его производные — цитраль и лилиаль[60].
Ацетальдегид используется для синтеза уксусной кислоты, этилового спирта, бутадиена для получения производных пиридина, пентаэритрита и кротонового альдегида, а также при синтезе поливинилацетата и пластмасс.
Альдегиды применяют для синтеза спиртов (бутиловых, 2-этилгексанола, пентаэритрита), карбоновых кислот, полимеров, антиоксидантов, пиридиновых оснований[1].
Примечания
- Кнунянц И. Л. и др. т.1 А-Дарзана // Химическая энциклопедия. — М.: Советская энциклопедия, 1988. — С. 196-198. — 623 с. — 100 000 экз.
- IUPAC Gold Book — aldehydes. Дата обращения: 7 июля 2013. Архивировано 9 июля 2013 года.
- Liebig J. Sur les Produits de l'Oxidation de l'Alcool (фр.) // Annales de chimie et de physique. — 1835. — Vol. 59. — P. 290.
- Senning A. Elsevier's Dictionary of Chemoetymology. — Elsevier, 2007. — P. 151. — ISBN 978-0-444-52239-9.
- Леенсон И. А. Откуда твоё имя? Статья шестая. Органические соединения. Дата обращения: 25 июня 2013. Архивировано 29 июня 2013 года.
- IUPAC Nomenclature of Organic Chemistry (англ.). ACD/Labs. Дата обращения: 24 августа 2009. Архивировано 21 августа 2011 года.
- Кан Р., Дермер О. Введение в химическую номенклатуру = Introduction to Chemical Nomenclature / Пер. с англ. Н. Н. Щербиновской, под ред. В. М. Потапова, Р. А. Лидина. — М.: Химия, 1983. — С. 139—140.
- Справочник химика / Редколлегия: Никольский Б. П. и др.. — 2-е издание. — Ленинград, Москва: Химия, 1964. — Т. 2. — С. 270, 285, 295.
- Петров А. А., Бальян Х. В., Трощенко А. Т. Органическая химия. — Иван Федоров, 1981. — Т. 1. — С. 165-184. — 672 с. — ISBN 5-81940-067-4.
- Несмеянов А.Н., Несмеянов Н.А. Начала органической химии. М., ,. Начала органической химии. — Химия, 1974.
- Реутов, 2004, т. 2, с. 265—273.
- Десс-Мартина Реагент (Dess Martin Periodinane). www.khimia.ru. Дата обращения: 21 июля 2016.
- Реутов, 2004, т. 3, с. 12—13.
- Шабаров Ю.С. Органическая химия. — Лань, 2011. — С. 218-221. — 848 с.
- Реутов, 2004, т. 1, с. 480—483.
- Реутов, 2004, т. 3, с. 13—14.
- Реутов, 2004, т. 3, с. 203—205.
- Реутов, 2004, т. 3, с. 235.
- Марч Дж. Органическая химия. — М.: Мир, 1987. — Т. Т. 2. — С. 359—363.
- Реутов, 2004, т. 3, с. 16—20.
- Kohlpaintner и др., 2013, p. 6—8.
- Reuss G., Disteldorf W., Gamer A. O., Hilt A. Formaldehyde // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2000. — doi:10.1002/14356007.a11_619.
- Eckert M., Fleischmann G., Jira R., Bolt H. M., Golka K. Acetaldehyde // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2006. — doi:10.1002/14356007.a01_031.pub2.
- Brühne F., Wright E. Benzaldehyde // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2011. — doi:10.1002/14356007.a03_463.pub2.
- Kohlpaintner и др., 2013, p. 2—3.
- Реутов, 2004, т. 3, с. 10—11.
- Kohlpaintner и др., 2013, p. 3, 13.
- Реутов, 2004, т. 3, с. 23.
- Реутов, 2004, т. 3, с. 23—26.
- Реутов, 2004, т. 3, с. 27—28.
- Реутов, 2004, т. 3, с. 29—37.
- Реутов, 2004, т. 3, с. 38—39.
- Реутов, 2004, т. 3, с. 40—42.
- Реутов, 2004, т. 3, с. 44—49.
- Реутов, 2004, т. 3, с. 59—60.
- Реутов, 2004, т. 3, с. 60—62.
- Реутов, 2004, т. 3, с. 62—64.
- Реутов, 2004, т. 3, с. 64—65.
- Реутов, 2004, т. 3, с. 86.
- March, J. «Organic Chemistry: Reactions, Mechanisms, and Structures» J. Wiley, New York: 1992. ISBN 0-471-58148-8.
- Реутов, 2004, т. 3, с. 95—99.
- Реутов, 2004, т. 3, с. 124—134.
- Реутов, 2004, т. 3, с. 75—76.
- Реутов, 2004, т. 3, с. 79—81.
- Реутов, 2004, т. 3, с. 69—75.
- Реутов, 2004.
- Реутов, 2004, т. 3, с. 82—83.
- Реутов, 2004, т. 3, с. 49—57.
- Реутов, 2004, т. 3, с. 57—59.
- Реутов, 2004, т. 3, с. 84—85.
- Цветков Л.А. § 26. Альдегиды // Органическая химия. Учебник для 10 класса. — 20-е изд. — М.: Просвещение, 1981. — С. 120—129.
- Качественная реакция на альдегиды с гидроксидом меди (II) Архивная копия от 24 декабря 2014 на Wayback Machine — видеоопыт в Единой коллекции цифровых образовательных ресурсов
- Качественная реакция на альдегиды с фуксинсернистой кислотой Архивировано 24 декабря 2014 года. — видеоопыт в Единой коллекции цифровых образовательных ресурсов
- Пассет Б. В., Антипов М. А. Практикум по техническому анализу и контролю в производстве химико-фармацевтических препаратов и антибиотиков. — Медицина, 1981. — 272 с.
- Dr. Neil Glagovich. Fragmentation - Aldehydes. Архивировано 6 июля 2013 года.
- Н.С.Вульфсон, В.Г.Заикин,А.И.Микая. Масс-спектроскопия органических соединений. — Химия, 1986. — С. 197—198.
- Dr. Ian Hunt. Spectroscopic Analysis of Aldehydes (англ.). Department of Chemistry University of Calgary. Архивировано 6 июля 2013 года.
- Общая токсикология / под ред. А. О. Лойта. СПб.: ЭЛБИ-СПб., 2006
- Альдегиды (недоступная ссылка). Дата обращения: 27 июня 2013. Архивировано 29 июня 2013 года.
- G. Reuss, W. Disteldorf, A. O. Gamer, A. Hilt. "Formaldehyde" in Ullmann's Encyclopedia of Industrial Chemistry (англ.). — 2005. — doi:10.1002/14356007.a11 619.
Литература
- Кнунянц И. Л. и др. т.1 А-Дарзана // Химическая энциклопедия. — М.: Советская энциклопедия, 1988. — 623 с. — 100 000 экз.
- Реутов О. А. и др. Органическая химия. В 4 частях. — М.: Бином. Лаборатория знаний, 2004.
- Kohlpaintner C., Schulte M., Falbe J., Lappe P., Weber J., Frey G. D. Aldehydes, Araliphatic // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2013. — doi:10.1002/14356007.m01_m03.pub2.
- Kohlpaintner C., Schulte M., Falbe J., Lappe P., Weber J., Frey G. D. Aldehydes, Aliphatic // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2013. — doi:10.1002/14356007.a01_321.pub3.
| В библиографических каталогах |
|---|
| Углеводороды | |
|---|---|
| Кислородсодержащие | |
| Азотсодержащие | |
| Серосодержащие | |
| Фосфорсодержащие | |
| Галогенорганические | |
| Кремнийорганические | |
| Элементоорганические | |
| Другие важные классы | |