Амиды
Ами́ды — производные кислородсодержащих кислот (карбоновых либо минеральных), в которых гидроксильная группа кислотного остатка заменена аминогруппой (незамещённой или замещённой). Амиды также можно рассматривать как ацилпроизводные аминов. Соединения с одним, двумя или тремя ацильными заместителями у атома азота называются первичными, вторичными и третичными амидами соответственно. Вторичные амиды также называют имидами[1].
- Это статья об амидах кислот. О неорганических веществах, содержащих ион NH2-, смотрите в статье Амиды металлов.
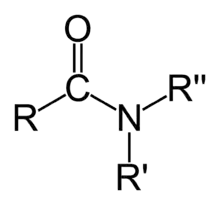
Амиды карбоновых кислот — карбоксамиды RCO−NR1R2 (где R1 и R2 — водород или другой остаток) — обычно называют просто амидами. В случае же других кислот, в соответствии с рекомендациями IUPAC, при именовании амида в качестве префикса указывается название класса кислоты, например амиды сульфокислот RSO2NH2 называются сульфамидами. Аналоги амидов, формально являющиеся продуктами замещения кислорода на халькоген, называются тиоамидами, селеноамидами и теллуроамидами[1].
Номенклатура
Название класса амидов происходит от названия аммиака. Золотая книга ИЮПАК делит амиды на первичные, вторичные и третичные в зависимости от числа ацильных остатков при атоме азота (ацильная группа — фрагмент, содержащий карбонильную (>C=O) и алкильную (R) группы, то есть фрагмент вида R—C=O). Однако ту же классификацию применяют и в тех случаях, если остатки не ацильные, а любые органические:
Если атом азота содержит две или три ацильные группы, такие соединения называют также имидами и триациламинами соответственно. Циклические амиды называют лактамами[2].
Названия первичных амидов образуют от названий соответствующих карбоновых кислот, добавляя к ним «-амид»:
Если атом азота дополнительно замещён, его заместители перечисляют в начале названия с приставкой «N» вместо локанта (N,N-диметилформамид)[2]. Моноамиды дикарбоновых кислот называют с помощью окончания «-амовая кислота», например моноамид фталевой кислоты можно назвать фталамовой кислотой[3].
Строение и физические свойства
Физические свойства амидов
Первичные и вторичные амиды представляют собой кристаллические вещества (за исключением жидких формамида и N-метилформамида); третичные амиды — жидкости. В воде хорошо растворяются лишь низшие алифатические амиды[2].
Строение амидной группы
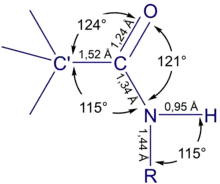
Строение амидной группы изучалось методом рентгеноструктурного анализа. Было показано, что в кристаллических амидах оно примерно одинаково. Амидная группа является плоской, атомы С', C, O, N лежат в одной плоскости, но атомы водорода при атоме азота не лежат в этой плоскости. Углы вокруг карбонильного атома углерода имеют следующие значения: α(С'-C=O) = 124°, α(С'-C-N) = 115°, α(N-C=O) = 121°. Длины связей равны: l(C'-C) = 1,52 Å, l(C=O) = 1,24 Å, l(C-N) = 1,34 Å. Определён также угол α(H-N-R) у вторичных амидов: он составляет 115°. Длины связей при атоме азота равны: l(N-H) = 0,95 Å, l(N-R) = 1,44 Å. В газовой фазе у амидов чуть укорочена связь C=O (с 1,24 до 1,19-1,21 Å) и удлинена связь C-N (с 1,34 до 1,36-1,37 Å); углы остаются примерно теми же[4].
Исследование ряда амидов как в твёрдой, так и в газовой фазе показало, что существует обратная связь между длинами связей C=O и С-N: при увеличении одной вторая укорачивается. Связывают это с тем, что атом азота в амидной группе находится в сопряжении с карбонильной группой и в реальную структуру вносят вклад две резонансные структуры А и В. С увеличением вклада полярной структуры В длина связи C=O увеличивается, а длина связи C-N уменьшается[5].

Изомерия амидов
Из-за наличия системы сопряжения связь C-N имеет частично двоевязный характер, поэтому вращение вокруг неё затруднено и амиды могут существовать в виде цис/транс-изомеров. У вторичных амидов сильно преобладают транс-изомеры: небольшую долю цис-изомеров удалось обнаружить только в том случае, если группа R при карбонильном атоме углерода — это водород. Даже такой объёмный заместитель R' как трет-бутил предпочитал находиться рядом с кислородом, чем с менее объёмным водородом. У третичных амидов преимущественная конфигурация более очевидно связана со пространственными затруднениями[6].
Спектроскопические характеристики
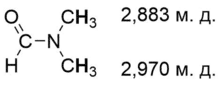
В спектрах ЯМР протоны амидной группы дают сигнал в области 5—8 м. д.[2]. Поскольку вращение вокруг амидной связи затруднено, два атома водорода при атоме азота в амидах типа RCONH2 дают два отдельных сигнала, не усредняясь даже при температурах выше комнатной. Например, в формамиде HCONH2 протон, находящийся в цис-положении к атому кислорода группы С=О, является более экранированным. В N,N-диалкиламиды две алкильные группы также имеют различное магнитное окружение и дают отдельные сигналы. Например, в диметилформамиде HCONMe2 более экранированной является также метильная группа, более близкая к атому кислорода. Химический сдвиг этих заместителей сильно зависит от растворителя; в ароматических растворителях их положение может меняться[8].
В ИК-спектрах амиды характеризуются двумя типами колебаний: колебаниями связи N-H выше 3000 см–1 и колебаниями связи C=O в области 1700—1600 см–1[2].
| Соединение | Колебание N-H | Колебание C=O |
|---|---|---|
| первичные амиды | две полосы при 3500—3400 см–1 | две полосы: при 1690—1630 см–1 (амидная полоса I) и при 1620—1590 см–1 (амидная полоса II) |
| вторичные амиды | одна полоса при 3460—3420 см–1 | две полосы: при 1690—1630 см–1 (амидная полоса I) и при 1550—1510 см–1 (амидная полоса II) |
| третичные амиды | — | одна полоса при 1670—1630 см–1 (амидная полоса І) |
Получение
Из карбоновых кислот и аминов
Амиды образуются при нагревании карбоновых кислот с аммиаком или первичными и вторичными аминами. Этот процесс возможен благодаря тому, что аммиак и амины являются более сильными нуклеофилами, чем вода и спирты. Пространственно затруднённые карбоновые в эту реакцию не вступают, хотя некоторые промышленно важные амиды, например сукцинимид и фталимид, получают именно так[9].

Наиболее вероятным механизмом этого превращения является механизм присоединения — отщепления, поскольку по такому механизму протекает обратная реакция гидролиза амидов. В случае дикарбоновых кислот промежуточно образуются их ангидриды. Амиды нельзя проацилировать простым нагреванием с карбоновой кислотой, поскольку при этом происходит реакция переацилирования[10]. Для аминокислот эта реакция осуществляется внутримолекулярно: при этом они превращаются в циклические амиды — лактамы. Например, при нагревании до 180 °С гамма-аминомасляная кислота даёт гамма-бутиролактам[9].
Реакциями ацилирования
Универсальным методом получения амидов является ацилирование аммиака, первичных и вторичных аминов хлорангидридами и ангидридами карбоновых кислот, кетенами и сложными эфирами[9].

Большинство таких реакций протекает по механизму присоединения — отщепления. Согласно этому механизму, чем выше частичный положительный заряд на карбонильном атоме углерода, тем выше скорость реакции. Соответственно, ацилирующие реагенты можно расположить в ряд активности: RCOR < RCONR2 < RCOOR < (RCO)2O < RCOHal < RCOBF4. Также скорость реакции зависит от нуклеофильности амина, которую условно можно связать с основностью амина: алкиламины > ариламины > амиды. Внутримолекулярное ацилирование происходит легче, чем межмолекулярное[11].
В реакции с хлорангидридами происходит выделение хлороводорода, поэтому в реакцию необходимо брать двойное количество амина, чтобы второй эквивалент связал этот хлороводород. Образующаяся аммониевая соль выпадает в осадок и фильтруется. Как следствие, максимальный выход амида из амина составляет 50 %. Как вариант, можно использовать другие органические и неорганические основания, чтобы повысить выход. Например, в реакции Шоттена — Баумана используется гидроксид натрия или гидроксид калия. Из органических оснований применяются пиридин, диметиланилин, триэтиламин и др.[12]
Из нитрилов
Нитрилы можно в контролируемых условиях гидролизовать до амидов. Гидролиз протекает как в кислой, так и в щелочной среде. Например, ароматические амиды и амиды затруднённых карбоновых кислот получают гидролизом серной кислотой, поскольку для них стадия гидролиза до карбоновой кислоты протекает медленно. Для многих же незатрудённых нитрилов этот способ не подходит, потому что амид легко гидролизуется дальше. Также в качестве кислотных катализаторов используют соляную кислоту, фосфорную кислоту и трифторид бора[13].


Основный катализ для этих реакций использовался реже из-за дальнейшего гидролиза амида. Надёжным способом из этой группы является использование щелочного раствора пероксида водорода[13].
Также нитрилы можно превратить во вторичные амиды по реакции Риттера. В этом случае нитрилы алкилируются по атому азота карбокатионами, полученными из алкенов и концентрированной серной кислоты, давая вторичный амид[9]. Эту реакцию можно провести, заменив алкены на соответствующие спирты[14].
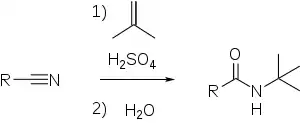
Алкены и спирты, дающие в кислой среде вторичные карбокатионы, вступают в реакцию Риттера медленнее, чем те, которые дают третичные карбокатионы. Первичные спирты ввести в эту реакцию можно только в очень жёстких условиях. Также для генерирования карбокатионов использовали разветвлённые алканы и циклоалканы, алкилгалогениды и карбоновые кислоты[14].
Перегруппировки
В промышленности используется метод получения амидов через перегруппировку Бекмана. В этой реакции оксимы вводят в реакцию с минеральными кислотами, кислотами Льюиса или полифосфорной кислотой. В лабораторных условиях этот метод не применяется, однако на практике он полезен для перегруппировки жирноароматических кетонов, доступных благодаря реакции ацилирования по Фриделю — Крафтсу, а также для получения лактамов[9][15].
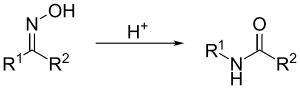
Существует ряд других перегруппировок, которые также приводят к амидам. Среди них перегруппировка Шмидта, реакция Вильгеродта и др.[15]
Химические свойства
Гидролиз
При действии горячей воды или водного пара амиды гидролизуются. Реакция протекает медленно, поскольку вода обладает низкой реакционной способностью по отношению к амидам: некоторые амиды даже перекристаллизовывают из неё. Гидролизу способствует щелочная либо кислая среда: в первом случае атаку на амид осуществляет более нуклеофильный гидроксид-ион OH-, а во втором случае амид протонируется по атому кислорода, из-за чего амидная группа становится более восприимчивой к атаке[16].
Почти все амиды гидролизуются в щелочной среде. Реакция ускоряется, если в амиде присутствуют электроноакцепторные группы, и замедляется при наличии пространственных затруднений. Катализ может осуществляться не обязательно протоном или гидроксид-ионом: возможен и общий кислотно-основный катализ[17][18].
Восстановление
Амиды устойчивы к восстановлению, и лишь сильные восстановители (гидриды, натрий в жидком аммиаке и электролиз) вступают с ними в реакции. При обработке первичных амидов алюмогидридом лития в диэтиловом эфире или тетрагидрофуране они восстанавливаются до первичных аминов. Аналогично, вторичные амиды дают вторичные амины, а третичные амиды — третичные амины. Лактамы в этих условиях дают циклические амины. Также в этих реакциях можно использовать диборан и электролиз[19][20].

N,N-Диалкиламиды можно контролируемо восстановить до альдегидов: в этом случае вместо алюмогидрида лития удобнее использовать более слабый восстановитель триэтоксиалюмогидрид лития LiAlH(OEt)3, получаемый из LiAlH4 и этанола. Также такое восстановление можно провести под действием гидрида диизобутилалюминия и натрия в жидком аммиаке[19][20].

Дегидратация
Первичные амиды отщепляют воду, давая нитрилы, под действием хлористого тионила, оксалилхлорида и оксихлорида фосфора. Наилучшим вариантом считается использование хлористого тионила в ДМФА. Также с этой целью используют трифторуксусный ангидрид в присутствии пиридина[21].
Нитрозирование
Первичные амиды легко разлагаются на холоде раствором азотистой кислоты, при этом выделяется азот и образуется соответствующая карбоновая кислота. В практическом плане преимущество имеют алкилнитриты RONO и тетрафторборат нитрозония NO+BF4-. Вторичные амиды, по аналогии с аминами, дают N-нитрозоамиды[22][23].

Галогенирование и перегруппировка Гофмана
Первичные и вторичные амиды реагируют с NaOCl, NaOBr, давая соответствующие N-хлорамиды или N-бромамиды. Сами эти соединения являются селективными галогенирующими реагентами (типичным примером является N-бромсукцинимид). Также N-галогенамиды в избытке щёлочи вступают в перегруппировку Гофмана, отщепляя при этом молекулу CO2 и давая амин, имеющий на один атом углерода меньше, чем исходный амид[24].
Реакции с металлоорганическими реагентами
Металлоорганические соединения, такие как реактивы Гриньяра и алкиллитиевые реагенты, могут присоединяться к карбонильной группе амида. Такие реакции с первичными и вторичными амидами бесполезны, поскольку реагент взаимодействует с кислым протоном при атоме азота и превращается в соответствующий алкан. Третичные амиды дают с реактивами Гриньяра прочные продукты присоединения, которые при обработке водной кислотой дают кетоны. Диметилформамид даёт соответствующий альдегид[25].

При использовании избытка реактива Гриньяра происходит формальное замещение карбонильного атома кислорода двумя алкильными группами[25].

Кислотно-основные свойства
Амиды обладают очень слабо выраженными кислотными и основными свойствами. Реагируя со щелочными металлами, они дают соли, легко разлагаемые водой. Некоторые соли тем не менее устойчивы (ртутная соль ацетамида используется при протравке зерна). Амиды способны присоединять протон в присутствии сильной кислоты, образуя соли.
Применение
В промышленности амиды используются в качестве пластификаторов бумаги и искусственной кожи, для экстракции радиоактивных металлов, в качестве исходных соединений для синтеза полимеров, как промежуточные продукты в производстве красителей и сульфамидных препаратов[2].
Примечания
- IUPAC Gold Book — amides. Дата обращения: 2 апреля 2019.
- Химическая энциклопедия, 1988, с. 127.
- Кан Р., Дермер О. Введение в химическую номенклатуру / Пер. с англ. Н. Н. Щербиновской, под ред. В. М. Потапова и Р. А. Лидина. — М. : Химия, 1983. — С. 136.
- Zabitsky, 1970, p. 2–3.
- Zabitsky, 1970, p. 6.
- Zabitsky, 1970, p. 19–20.
- Spectral Database for Organic Compounds, SDBS. Дата обращения: 3 апреля 2019.
- Zabitsky, 1970, p. 14–18.
- Реутов, 2004, с. 237–239.
- Zabitsky, 1970, p. 106–108.
- Zabitsky, 1970, p. 74–77.
- Zabitsky, 1970, p. 77–81.
- Zabitsky, 1970, p. 119–122.
- Zabitsky, 1970, p. 125–129.
- Zabitsky, 1970, p. 131–132.
- Zabitsky, 1970, p. 816.
- Реутов, 2004, с. 240–242.
- Zabitsky, 1970, p. 824.
- Реутов, 2004, с. 242–244.
- Zabitsky, 1970, с. 795–801.
- Реутов, 2004, с. 244–245.
- Реутов, 2004, с. 245.
- Zabitsky, 1970, с. 780–784.
- Реутов, 2004, с. 246–247.
- Zabitsky, 1970, p. 847.
Литература
- Садовая Н. К. Амиды карбоновых кислот // Химическая энциклопедия : в 5 т. / Гл. ред. И. Л. Кнунянц. — М.: Советская энциклопедия, 1988. — Т. 1: А—Дарзана. — С. 127–128. — 623 с. — 100 000 экз. — ISBN 5-85270-008-8.
- Реутов О. А., Курц А. Л., Бутин К. П. Органическая химия : в 4 т.. — 4-е изд. — М. : БИНОМ. Лаборатория знаний, 2004. — Т. 3, 18.9 Амиды карбоновых кислот. — С. 237–252. — 750 экз. — ISBN 978-5-9963-1335-8.
- The Chemistry of Amides : [англ.] / Ed. Jacob Zabitsky. — Interscience Publishers, 1970. — ISBN 0471980498.
Ссылки
- Амиды // Энциклопедический словарь Брокгауза и Ефрона : в 86 т. (82 т. и 4 доп.). — СПб., 1890—1907.
| Углеводороды | |
|---|---|
| Кислородсодержащие | |
| Азотсодержащие | |
| Серосодержащие | |
| Фосфорсодержащие | |
| Галогенорганические | |
| Кремнийорганические | |
| Элементоорганические | |
| Другие важные классы | |
