Алкены
Алке́ны (олефи́ны, этиле́новые углеводоро́ды) — ациклические непредельные углеводороды, содержащие одну двойную связь между атомами углерода, образующие гомологический ряд с общей формулой CnH2n.
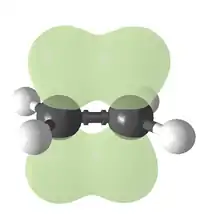
Атомы углерода при двойной связи находятся в состоянии sp2-гибридизации и имеют валентный угол 120°. Простейшим алкеном является этилен (C2H4). По номенклатуре IUPAC, названия алкенов образуются от названий соответствующих алканов заменой суффикса «-ан» на «-ен»; положение двойной связи указывается арабской цифрой.
Углеводородные радикалы, образованные от алкенов, имеют суффикс «-ени́л». Тривиальные названия: CH2=CH— «вини́л», CH2=CH—CH2— «алли́л».
Гомологический ряд и изомерия
Алкены, число атомов углерода в которых больше двух, (то есть кроме этилена) имеют свои изомеры. Для алкенов характерны изомерия углеродного скелета, положения двойной связи, межклассовая и геометрическая. Например, единственным изомером пропилена является циклопропан (C3H6) по межклассовой изомерии. Начиная с бутилена, существуют изомеры по положению двойной связи (бутен-1 и бутен-2), по углеродному скелету (изобутилен или метилпропилен) и геометрические изомеры (цис-бутен-2 и транс-бутен-2). С ростом числа атомов углерода в молекуле количество изомеров быстро возрастает.

Гомологический ряд алкенов:
| Этен (этилен) | C2H4 |
| Пропен (пропилен) | C3H6 |
| Бутен (бутилен) | C4H8 |
| Пентен | C5H10 |
| Гексен | C6H12 |
| Гептен | C7H14 |
| Октен | C8H16 |
| нонен | C9H18 |
| децен | C10H20 |
Алкены могут существовать в виде пространственных или геометрических изомеров.
Различают:
- цис- изомеры: заместители расположены по одну сторону от двойной связи;
- транс- изомеры: заместители расположены по разные стороны от двойной связи.
IUPAC рекомендует называть геометрические изомеры по следующей номенклатуре:
- Z- изомеры: старшие заместители у углеродных атомов двойной связи находятся по одну сторону относительно двойной связи;
- E- изомеры: старшие заместители у углеродных атомов двойной связи находятся по разные стороны относительно двойной связи.

Электронное строение двойной связи
В соответствии с теорией гибридизации двойная связь образуется за счёт перекрывания вдоль линии связи С-С sp2-гибридных орбиталей атомов углерода (σ-связь) и бокового перекрывания углеродных p-орбиталей (π-связь).

В состоянии sp2-гибридизации электронное состояние атома углерода можно представить следующим образом:

Все атомы этилена лежат в одной плоскости, а величина валентного угла связи C-H практически равна 120 °. Центры углеродных атомов в этилене находятся на расстоянии 0,134 нм, то есть длина двойной связи несколько короче, чем С-С.
Согласно теории молекулярных орбиталей линейная комбинация двух атомных 2p-орбиталей углерода формирует две молекулярные π-орбитали этилена[1]:

Первый потенциал ионизации этилена составляет 10,51 эВ[2], что позволяет электрону относительно легко уходить (электрофильное взаимодействие) с высшей занятой молекулярной орбитали (ВЗМО). В то же время, низшая связывающая молекулярная орбиталь (НСМО) этилена имеет достаточно низкую энергию: −1,6-1,8 эВ, что объясняет относительную лёгкость присоединения электрона с образованием аниона[2] (нуклеофильное взаимодействие).
Добавление метильного заместителя снижает потенциал ионизации π- электронов примерно на 0,6-0,8 эВ и повышает энергию НСМО на 0,2 эВ, а ВЗМО на 0,7 эВ[2].
История открытия
Впервые этилен был получен в 1669 году немецким химиком и врачом И.И. Бехером действием серной кислоты на этиловый спирт. Учёный установил, что его «воздух» более химически активен, чем метан, однако идентифицировать полученный газ он не смог и названия ему не присвоил[3].
Вторично и тем же способом «воздух Бехера» был получен и описан голландскими химиками Я.Р. Дейманом, Потс-ван-Трооствиком, Бондом и Лауверенбургом в 1795 году. Они назвали его «маслородным газом» так как при взаимодействии с хлором, он образовывал маслянистую жидкость — дихлорэтан (об этом стало известно позднее). По-французски «маслородный» — oléfiant. Французский химик Антуан Фуркруа ввёл этот термин в практику, а когда были обнаружены другие углеводороды такого же типа, это название стало общим для всего класса олефинов (или, по современной номенклатуре, алкенов)[4].
В начале XIX века французский химик Ж. Гей-Люссак обнаружил, что этанол состоит из «маслородного» газа и воды. Этот же газ он обнаружил и в хлористом этиле[5]. В 1828 году Ж. Дюма и П. Буллей предположили, что этилен представляет собой основание, способное давать соли подобно аммиаку. Якоб Берцелиус принял эту идею, назвав соединение «этерином» и обозначив буквой E[6].
Определив, что этилен состоит из водорода и углерода, долгое время химики не могли выписать его настоящую формулу. В 1848 году Кольбе писал формулу этилена как С4Н4, этого же мнения придерживался и Либих. Ж. Дюма правильно определил состав вещества, но его структура по-прежнему была описана неверно: С2НН3[5].
В 1862 году немецкий химик-органик Э.Эрленмейер предположил наличие в молекуле этилена двойной связи, а в 1870 году известный российский учёный А. М. Бутлеров признал эту точку зрения правильной, подтвердив её природу экспериментально[7].
Нахождение в природе и физиологическая роль алкенов
В природе ациклические алкены практически не встречаются[8]. Простейший представитель этого класса органических соединений — этилен (C2H4) — является гормоном для растений и в незначительном количестве в них синтезируется.
Один из немногих природных алкенов — мускалур (цис- трикозен-9) является половым аттрактантом самки домашней мухи (Musca domestica).
![]()
Низшие алкены в высоких концентрациях обладают наркотическим эффектом. Высшие члены ряда также вызывают судороги и раздражение слизистых оболочек дыхательных путей[9].
Отдельные представители:
- Этилен — вызывает наркоз, обладает раздражающим и мутагенным действием.
- Пропилен — вызывает наркоз (сильнее, чем этилен), оказывает общетоксическое и мутагенное действие.
- Бутен-2 — вызывает наркоз, обладает раздражающим действием[9].
Физические свойства
- Температуры плавления и кипения алкенов (упрощённо) увеличиваются с молекулярной массой и длиной главной углеродной цепи.
- При нормальных условиях алкены с C2H4 до C4H8 — газы; с пентена C5H10 до гептадецена C17H34 включительно — жидкости, а начиная с октадецена C18H36 — твёрдые вещества. Алкены не растворяются в воде, но хорошо растворяются в органических растворителях.
| Физические свойства алкенов[10] | |||||
|---|---|---|---|---|---|
| № | Название | Формула | Т плавления, ° C | Т кипения, ° C | Плотность, d20 4 |
| 1 | Этилен | С2H4 | −169,1 | −103,7 | 0,5700* |
| 2 | Пропилен | C3H6 | −187,6 | −47,7 | 0,5193* |
| 3 | Бутен-1 | C4H8 | −185,3 | −6,3 | 0,5951* |
| 4 | цис-Бутен-2 | CH3-CH=CH-CH3 | −138,9 | 3,7 | 0,6213 |
| 5 | транс-Бутен-2 | CH3-CH=CH-CH3 | −105,5 | 0,9 | 0,6042 |
| 6 | 2-Метилпропен-1 | CH3-C(CH3)=CH2 | −140,4 | −7,0 | 0,5942* |
| 7 | Пентен-1 | CH2=CH-CH2-CH2-CH3 | −165,2 | 30,1 | 0,6405 |
| 8 | Гексен-1 | CH2=CH-CH2-CH2-CH2-CH3 | −139,8 | 63,5 | 0,6730 |
| 9 | Гептен-1 | С7H14 | −119,0 | 93,6 | 0,6970 |
| 10 | Октен-1 | С8H16 | −101,7 | 121,3 | 0,7140 |
| … | Гептадецен[11] | С17H34 | 4,1 | 284,4 | 0,7811 |
* Значения измерены при температуре кипения.
Химические свойства
Алкены химически активны. Их химические свойства во многом определяются наличием двойной связи. Для алкенов наиболее характерны реакции электрофильного присоединения и реакции радикального присоединения. Реакции нуклеофильного присоединения обычно требуют наличие сильного нуклеофила и для алкенов не типичны.
Особенностью алкенов являются также реакции циклоприсоединения и метатезиса.
Алкены легко вступают в реакции окисления, гидрируются с сильными восстановителями или водородом под действием катализаторов, а также способны к радикальному замещению.
Реакции электрофильного присоединения
В данных реакциях атакующей частицей является электрофил.
Галогенирование
Галогенирование алкенов, проходящее в отсутствие инициаторов радикальных реакций — типичная реакция электрофильного присоединения. Она проводится в среде неполярных инертных растворителей (например: CCl4):

Реакция галогенирования стереоспецифична —- присоединение происходит с противоположных сторон относительно плоскости молекулы алкена[1]
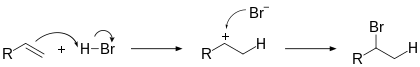
Механизм реакций подобного типа в общем виде:

Гидрогалогенирование
Электрофильное присоединение галогенводородов к алкенам происходит по правилу Марковникова:
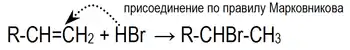
Однако в присутствии перекисей присоединение происходит преимущественно против этого правила (эффект Хараша)[1]:
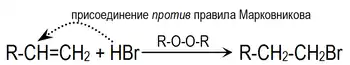
Это объясняется тем, что реакция в данном случае будет протекать по радикальному механизму и присоединение радикала Br. идёт по стерически наиболее доступному концевому атому углерода двойной связи:
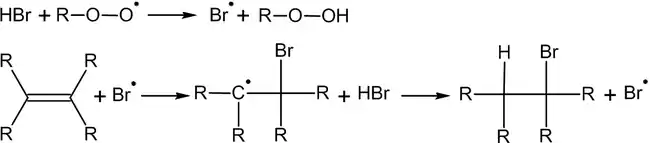
Гидроборирование
Присоединение гидридов бора к алкенам и последующее их расщепление в щелочной среде, открытое Г. Брауном в 1958 году, является столь важной реакцией, что за её обнаружение и изучение в 1979 году учёный был удостоен Нобелевской премии по химии[12].
Присоединение происходит многоступенчато с образованием промежуточного циклического активированного комплекса, причём присоединение бора происходит против правила Марковникова — к наиболее гидрогенизированному атому углерода:
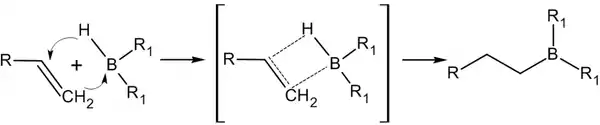
В синтезе используется, обычно, не собственно диборан, а его донорно-акцепторный комплекс с простым эфиром:
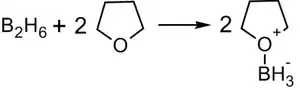
Алкилбораны легко расщепляются. Так под действием пероксида водорода в щелочной среде образуются спирты:
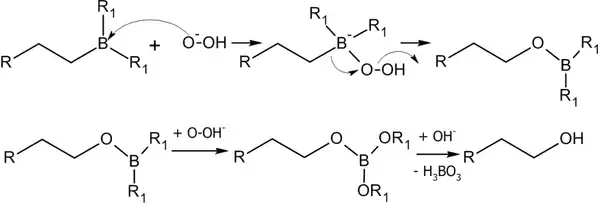
Реакция гидроборирования является реакцией син-присоединения — её результатом становятся цис-аддукты.
Гидратация
Реакция присоединения воды к алкенам протекает в присутствии серной кислоты[13]:

Реакция протекает по правилу Марковникова.
Алкилирование
Присоединение алканов к алкенам в присутствии кислотного катализатора (HF или H2SO4) при низких температурах приводит к образованию углеводорода с большей молекулярной массой и часто используется в промышленности[14]:

Данная реакция также может протекать по свободнорадикальному механизму в отсутствие катализатора при высокой температуре (500 °C) и давлении (15-30 МПа)[13].
Прочие реакции электрофильного присоединения
Для алкенов также характерны следующие реакции электрофильного присоединения[13]:
- Присоединение спирта с образованием простого эфира:

- Получение спиртов по реакции оксимеркурирования-демеркурирования:


- Присоединение хлорноватистой кислоты с образованием хлоргидринов:

- Присоединение хлорангидридов с дальнейшим получением ненасыщенных кетонов (реакция Кондакова, катализатор ZnCl2[15]):

Реакции радикального присоединения
В условиях, способствующих гомолитическому разрыву связи, (высокая температура, облучение, наличие свободных радикалов и пр.) присоединение к алкенам происходит по радикальному механизму[16].
по правилу Марковникова.



и т. п.
Механизм реакции:

Реакции присоединения карбенов
Карбены CR2: — высокореакционные короткоживущие частицы, которые способны легко присоединяться к двойной связи алкенов[17]. В результате реакции присоединения карбена образуются производные циклопропана:

Карбены в более характерном для них синглетном состоянии, вступая в реакцию, дают стереоспецифичные продукты син-присоединения[13].
Помимо собственно карбена, в подобные реакции могут вступать и его производные[13]:
и пр.

Часто реакции присоединения карбенов происходят без прямых доказательств их свободного присутствия, то есть происходит перенос карбена. Для этого случая, а также если генерация свободного карбена ставится под сомнение, пользуются термином карбеноид[18].
В лабораторной практике часто пользуются реакцией Симмонса — Смита[19]:

Подробнее о методах получения карбенов см. статью Карбены.
Гидрирование (реакция Сабатье — Сандеран)

Гидрирование алкенов непосредственно водородом происходит только в присутствии катализатора. Гетерогенными катализаторами гидрирования служат платина, палладий, никель [20].
Гидрирование можно проводить и в жидкой фазе с гомогенными катализаторами (например: катализатор Уилкинсона ((C6H5)3P)3Rh Cl)[20].
В качестве реагентов гидрирования могут выступать диимид (NH=NH), диборан (B2H6) и др[21].
Реакции радикального замещения
При высоких температурах (более 400 °C) реакции радикального присоединения, носящие обратимый характер, подавляются. В этом случае становится возможным провести замещение атома водорода, находящегося в аллильном положении при сохранении двойной связи:

Реакция носит радикальный характер и протекает аналогично хлорированию алканов.
Аллильное бромирование обычно проводят N-бромсукцинимидом (реакция Воля — Циглера)[22] в присутствии перекиси бензоила в среде тетрахлорметана или в бинарной смеси диметилсульфоксида и воды[20]:
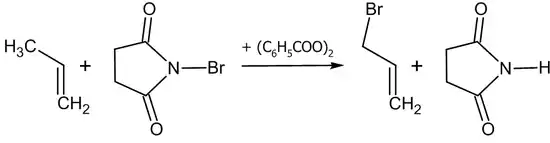
Окисление
Окисление алкенов может происходить в зависимости от условий и видов окислительных реагентов как с разрывом двойной связи, так и с сохранением углеродного скелета.
Окисление неорганическими окислителями
- В мягких условиях возможно окисление посредством присоединения по двойной связи двух гидроксильных групп[23]:

На первом этапе происходит присоединение оксида осмия к алкену, затем под действием восстановителя (Zn или NaHSO3) образовавшийся комплекс переходит к диолу (Реакция Криге).
Аналогично реакция идёт в нейтральной или слабощелочной среде под действием KMnO4 (Реакция Вагнера)[23]:

- При действии на алкены сильных окислителей (KMnO4 или K2Cr2O7 в среде Н2SO4) при нагревании происходит разрыв двойной связи:

(кетон)

- Некоторые окислители, например нитрат (III) таллия, окисляют алкены с перегруппировкой по следующей схеме[23]:

Окисление в присутствии солей палладия
В присутствии солей палладия этилен окисляется до ацетальдегида[1]:

Реакция идёт в кислой среде и является промышленным способом получения ацетальдегида.
Эпоксидирование
При действии на алкены пероксикарбоновых кислот образуются эпоксиды (реакция Прилежаева)[24]:
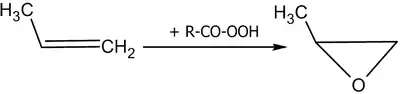
Реакция эпоксидирования используется для промышленного получения этиленоксида. Окислителем выступает кислород воздуха; процесс идёт на серебряном катализаторе при 200—250 °C под давлением.
Озонолиз
Озонолиз алкенов обычно проводят при низких температурах (от −80 до −30 °C) в инертном растворителе (гексан, тетрахлорметан, хлороформ, этилацетат и пр.). Непосредственные продукты озонолиза не выделяют, а подвергают дальнейшему гидролизу, окислению или восстановлению[23].
- Озонолиз в мягких условиях: алкен окисляется до альдегидов (в случае монозамещённых вицинальных углеродов), кетонов (в случае дизамещенных вицинальных углеродов) или смеси альдегида и кетона (в случае три-замещенного у двойной связи алкена).
На первой стадии происходит присоединение озона с образованием озонида. Далее под действием восстановителя (например: Zn + CH3COOH) озонид разлагается:
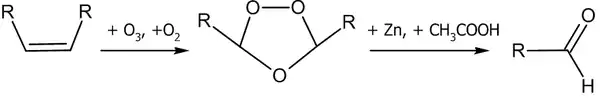
Если взять более сильный восстановитель, скажем — алюмогидрид лития, продуктом реакции будут спирты.
- Озонолиз в жёстких условиях — алкен окисляется до кислоты:

В данном случае разложение озонида происходит под действием окислителей (пероксид водорода, оксид серебра, пероксикислоты и пр.[23]).
Реакция карбонилирования
Алкены в присутствии катализатора, высокой температуры и давления присоединяют CO и H2 с образованием альдегидов[25]:

Аналогично протекает реакция CO и H2O с образованием карбоновых кислот[25] :

Если вместо воды использовать спирт, конечным продуктом реакции будет сложный эфир[25] :

Реакции полимеризации
Полимеризация алкенов может протекать как по свободнорадикальному, так и катионно-анионному механизму.
По первому методу получают полиэтилен высокого давления:

Катализатором реакции выступают пероксиды.
Второй метод предполагает использование в качестве катализаторов кислот (катионная полимеризация), металлорганических соединений (катализаторы Циглера-Натта, анионная полимеризация). Преимуществом метода является возможность получения стереоселективных полимеров.
Реакции свободнорадикального присоединения
Метатезис алкенов
Впервые данный тип реакций был обнаружен в середине прошлого века при изучении полимеризации этилена, а в затем был использован в 1966 году для промышленного синтеза бутена-2.
В 1967 году Н. Кальдерон, Х. Ю Чен и К. В. Скотт описали метатезис алкенов (в российской литературе часто употребляется термин реакция дисмутации алкенов, иначе говоря — реакцию обмена атомами при сохранении общей структуры алкена и его двойной связи) в условиях катализа хлоридом вольфрама (VI):

Реакция оказалась настолько важной в области практической препаративной химии, что исследовательская группа Роберта Граббса, разработавшая новый класс катализаторов (алкилиденовые комплексы рутения) метатезиса олефинов, получила в 2005 году Нобелевскую премию в области химии[26]. Эту премию также получили француз Ив Шовен в 1971 году, предложивший карбеновую теорию механизма реакции метатезиса[27], и американец Ричард Шрок, создавший в 1990 году первый металлорганический катализатор метатезиса алкенов[28].
В 2008 году польские химики продемонстрировали реакцию метатезиса в водном растворе с использованием коммерчески доступного рутениевого катализатора[29].
Технологические аспекты метатезиса алкенов рассмотрены в статье: Метатезис олефинов: современный путь к полипропилену
Методы получения алкенов
Основным промышленным методом получения алкенов является каталитический и высокотемпературный крекинг углеводородов нефти и природного газа. Для производства низших алкенов используют также реакцию дегидратации соответствующих спиртов.
В лабораторной практике обычно применяют метод дегидратации спиртов в присутствии сильных минеральных кислот[1], дегидрогалогенирование и дегалогенирование соответствующих галогенпроизводных; синтезы Гофмана, Чугаева, Виттига и Коупа[30].
Подробнее — см. соответствующие разделы ниже.
Дегидрирование алканов
Это один из промышленных способов получения алкенов[31][32]. Температура: 350—450 °C, катализатор — Cr2O3. Также используются алюмомолибденовые и алюмоплатиновые катализаторы[33]. Для получения транс-алкенов используют MOH/EtOH, для цис-производных NaNH2/NH3


Дегидрогалогенирование и дегалогенирование алканов
Отщепление галогенов у дигалогеналканов происходит в присутствии цинка[34]:

Дегидрогалогенирование проводят при нагревании действием спиртовыми растворами щелочей[35]:

При отщеплении галогенводорода образуется смесь изомеров, преобладающий из которых определяется правилом Зайцева: отщепление протона происходит от менее гидрогенизированного атома углерода.
Дегидратация спиртов
Дегидратацию спиртов ведут при повышенной температуре бани в присутствии сильных минеральных кислот[34]:


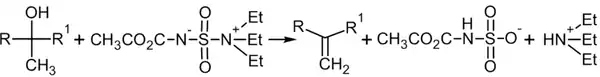
В современной практике алкены из вторичных и третичных спиртов также получают с использованием дегидратирующего реагента — реагента Бёрджесса[19]:
Гидрирование алкинов
Частичное гидрирование алкинов требует специальных условий и наличие катализатора (например, дезактивированного палладия — катализатора Линдлара)[34]:
(цис-изомер)

(транс-изомер)


Реакция Виттига
Реакция Виттига — стереоселективный синтез алкенов взаимодействием карбонильных соединений и алкилиденфосфоранов (илидов фосфониевых солей)[36]:



Для превращения солей фосфония в илиды используются бутиллитий, гидрид, амид или алкоголят натрия, а также некоторые другие сильные основания.
В реакцию могут вступать самые различные карбонильные соединения, среди которых ароматические и алифатические альдегиды и кетоны, в том числе содержащие двойные и тройные связи и различные функциональные группы.
В лабораторной практике часто используют более современную модификацию (1959 год) реакции Виттига — реакцию Хорнера-Уодсворта-Эммонса[37]:
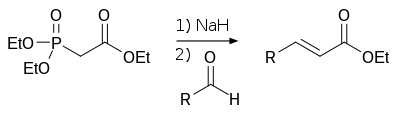
Преимущество использования фосфонатов заключается в том, что образующиеся в ходе реакции фосфаты легко отмываются водой. Кроме того, реакция позволяет избирать оптическое направление элиминирования, получая на выходе транс- (термодинамический контроль) или цис-изомеры (кинетический контроль)[19].
Реакция Кнёвенагеля
Реакция Кнёвенагеля — конденсация альдегидов или кетонов с соединениями, содержащими активную CH2-группу[19]:

Реакция имеет очень широкий диапазон применения, при этом помимо эфиров малоновой кислоты, в реакцию могут вступать и другие соединения, например: CH3CN, CH3NO2, LiCH2COOC2H5 и пр.[13].
Реакция Чугаева
Реакция Чугаева — взаимодействие спиртов с CS2 и NaOH с последующим метилированием и дальнейшим пиролизом образовавшихся S-метилксантогенатов[38]:


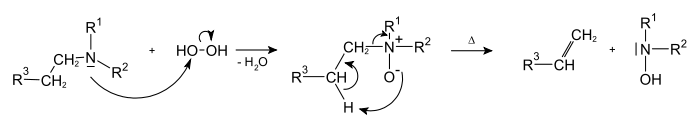
Реакция Гофмана
Исчерпывающее метилирование по Гофману — разложение четвертичных аммониевых оснований на алкен, третичный амин и воду[39]:

На первой стадии реакции действием метилиодида амин превращают в четвертичный аммонийиодид, который далее переводят в гидроксид действием оксида серебра, наконец, последний этап — разложение — ведут при 100—200 °C, часто при пониженном давлении[40].
Элиминирование по Гофману приводит к образованию наименее замещённых алкенов (против правила Зайцева).
Метод используется, в основном, для получения некоторых циклических алкенов и в химии алкалоидов[40].
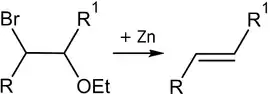
Реакция Бурда
Реакция Бурда — элиминирование брома и этоксигруппы из бромалкилэтиловых эфиров под действием цинковой пыли[41]:
Синтез из тозилгидразонов
Алкены можно получить разложением тозилгидразонов под действием оснований (Реакция Бэмфорда — Стивенса и Реакция Шапиро)[42]:
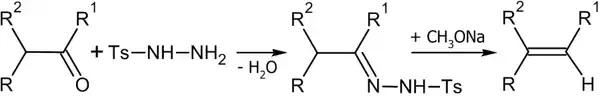
Реакция Бэмфорда — Стивенса и Реакция Шапиро протекают по одинаковому механизму. В первом случае используются натрий, метилат натрия, гидриды лития или натрия, амид натрия и т. п. Во втором: аллкиллитий и реактивы Гриньяра. В реакция Бэмфорда — Стивенса образуются более замещённые, а в реакция Шапиро — наименее замещённые алкены[43].
Реакция Перкина
Реакция Перкина — взаимодействие ароматических альдегидов с ангидридами карбоновых кислот в присутствии катализаторов основного характера (щелочных солей карбоновых кислот, третичных аминов и т. п.)[44]:
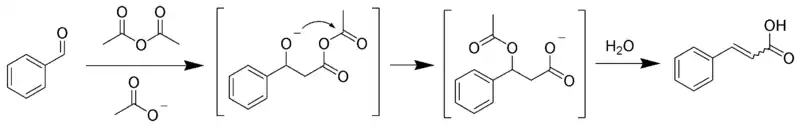
Последующим декарбоксилированием образующейся кислоты можно получить соответствующий алкен.
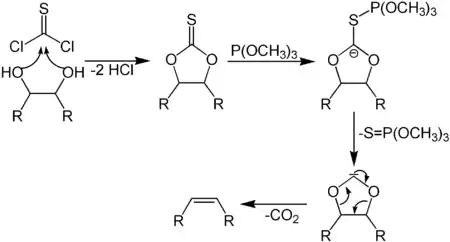
Синтез Кори — Винтера
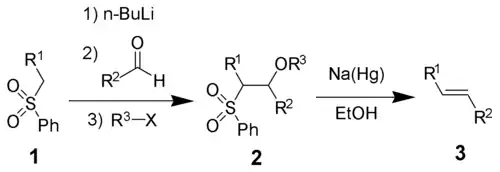
Олефинирование Жюлиа — Лижо
Идентификация алкенов
Химические методы идентификации алкенов
Часто для идентификации алкенов используют реакцию Вагнера: обесцвечивание раствора перманганата калия в слабощелочной среде (окисление алкенов до гликолей). Другой вариант — обесцвечивание раствора брома в четыреххлористом углероде при отсутствии выделения бромоводорода (реакция присоединения)[45].
Эти химические методы является очень общими, не селективными и не могут гарантированно определить алкены. Для подтверждения наличия двойной связи в соединении используют методы спектроскопии.
Масс-спектрометрические методы анализа алкенов
Масс-спектры алкенов по сравнению с алканами содержат более интенсивные M+ пики[46]. Существует эффективный экспресс-метод масс-спектрометрического исследования строения алкенов, заключающийся в изучении масс-спектров соответствующих алканов, образующихся при проведении газофазного гидрирования алкенов в токе водорода (кат. Pt, Pd) в микрореакторе, расположенном между газовым хроматографом и масс-спектрометром[47].
УФ-спектроскопические методы анализа алкенов
Алкены с изолированными двойными связями имеют интенсивную (ε от 6500 до 12000) широкую полосу поглощения, обусловленную переходом π→π, в области 165—200 нм. Наличие алкильных заместителей смещает эту полосу в длинноволновую область[48].
ИК-спектроскопические методы анализа алкенов
ИК-спектры алкенов имеют представленные в таблице характеристические полосы, вызванные валентными колебаниями связи С=С и C-H[49]:
| Типы колебаний и групп | Диапазон, см−1 | Примечание |
|---|---|---|
Валентные колебания связей C−H | ||
| R2C=CH2 | 3095-3075 | Могут наблюдаться мультиплеты |
| R2C=CHR | 3045-3010 | Дифференциация цис- и транс- изомеров невозможна |
Деформационные колебания связей C−H | ||
| RCH=CH2 | 990, 910 | |
| R,RC=CH2 | около 890 | |
| R,RC=CHR | 840-790 | |
| транс—RCH=CHR | около 950 | |
| цис−RCH=CHR | 730-665 | |
Валентные колебания связей C=С | ||
| транс−RCH=CHR | около 1675 | Полосы умеренной и высокой интенсивности, пригодные для идентификации ациклических и ненапряжённых систем |
| цис−RCH=CHR | около 1660 | |
| RCH=CR1R2 | около 1670 | |
| R2C=CH2 | около 1650 | |
| RCH=CH2 | около 1640 | |
| C=C−C=C | 1645-1600 | Положение полосы, более интенсивной чем у алкенов, зависит от геометрии сопряжённой системы |
| C=C−C=O | 1660-1580 | |
| C=C−(C=C)n | 1650-1580 | Полосы имеют мультиплетную структуру, а при больших n сливаются в одну широкую полосу |
| ArC=C | около 1630 | Положение полосы зависит от положения и природы заместителей |
ЯМР-спектроскопические методы анализа алкенов
ЯМР-спектроскопические методы анализа алкенов позволяют идентифицировать сигналы атомов водорода алкенов, тем самым получив важную информацию о структуре углеводородов. Эти сигналы лежат в диапазоне 4-8 м.д. Существует эмпирическая зависимость, позволяющая достаточно точно вычислить сдвиги протонов алкенов[50]:
δC=C-H = 5,25 + Zгем + Zцис + Zтранс
где Z-аддитивные параметры экранирования соответствующих заместителей.
Значения Z для отдельных заместителей представлены в таблице[50]:
| Заместитель | Zгем | Zцис | Zтранс |
|---|---|---|---|
| H | 0,00 | 0,00 | 0,00 |
| Алкил | 0,45 | -0,22 | -0,28 |
| Алкил (цикл.)* | 0,69 | -0,25 | -0,28 |
| CH2Ar | 1,05 | -0,29 | -0,32 |
| CH2X (X:F, Cl, Br) | 0,70 | 0,11 | -0,04 |
| CH2OH | 0,64 | -0,01 | -0,02 |
| CH2NH2 | 0,58 | -0,10 | -0,08 |
| C=C (изолир.) | 1,00 | -0,09 | -0,23 |
| C=C (сопряж.) | 1,24 | 0,02 | -0,05 |
| Ar | 1,38 | 0,36 | -0,07 |
| Cl | 1,08 | 0,18 | 0,13 |
| Br | 1,07 | 0,45 | 0,55 |
| OR | 1,22 | -1,07 | -1,21 |
| OC(O)R | 2,11 | -0,35 | -0,64 |
| CHO | 1,02 | 0,95 | 1,17 |
| COOH | 0,97 | 1,41 | 0,71 |
| COOR | 0,80 | 1,18 | 0,55 |
* — Двойная связь и алкил входят в цикл
Применение алкенов
Алкены являются важнейшим химическим сырьём.
Промышленное использование этилена
Этилен используется для производства целого ряда химических соединений: винилхлорида, стирола, этиленгликоля, этиленоксида, этаноламинов, этанола, диоксана, дихлорэтана, уксусного альдегида и уксусной кислоты[34]. Полимеризацией этилена и его прямых производных получают полиэтилен, поливинилацетат, поливинилхлорид, каучуки и смазочные масла.
Мировое производство этилена составляет порядка 100 млн тонн в год[51] (по данным на 2005 год: 107 млн тонн[52]).
Промышленное использование пропилена
Пропилен в промышленности применяется, в основном, для синтеза полипропилена (62 % процента всего выпускаемого объёма[53]). Также из него получают кумол, окись пропилена, акрилонитрил, изопропанол, глицерин, масляный альдегид[34].
В настоящее время мировые мощности по выпуску пропилена составляют около 70 млн тонн в год[53]. По прогнозам специалистов, потребность в пропилене в ближайшем будущем будет существенно превышать объёмы его производства, причём, ожидается, что к 2010 году объём его мирового выпуска достигнет 90 млн тонн[54].
Промышленное использование прочих алкенов
Бутилены применяют для производства бутадиена, изопрена, полиизобутилена, бутилкаучука, метилэтилкетона и пр[55].
Изобутилен — сырьё для получения бутилкаучука, изопрена, трет-бутанола; используется для алкилирования фенолов при синтезе ПАВ. Его сополимеры с бутенами применяют как присадки к маслам и герметики.
Высшие алкены С10−С18 применяют при синтезе ПАВ, а также для получения высших спиртов.
Дополнительные внешние источники
Общие лекции по химии алкенов
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 7 (Алкены. Строение, получение, реакционная способность.)
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 8 (Алкены. Реакционная способность.)
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 9 (Алкены. Реакционная способность.)
- Курц А. Л., Ливанцов М. В., Ливанцова Л. И. Алкены (Часть I). Химический факультет МГУ, 1998 год.
- Курц А. Л., Ливанцов М. В., Ливанцова Л. И. Алкены (Часть II). Химический факультет МГУ, 1999 год.
Учебная литература
- Нейланд О. Я. Глава II. Алкены // Органическая химия: Учеб. для хим. вузов. — М.: «Высшая школа», 1990. — С. 102—130. — ISBN 5-06-001471-1.
- Робертс Дж., Касерио М. Глава 6. Алкены. Структура, спектры и стереоизомерия. Глава 7. Алкены. Реакции двойных углерод-углеродных связей // Основы органической химии / Под редакцией академика Несмеянова А.Н.. — 2-е, дополненное. — М.: Мир, 1978. — Т. 1. — С. 171—235.
- Реутов О. А., Курц А. Л., Бутин К. П. Органическая химия. В 4 частях. — 3-е издание. — М.: Бином. Лаборатория знаний, 2007. — Т. 1. — 568 с. — ISBN 978-5-94774-613-6.
- Травень В. Ф. Глава 5. Алкены // Органическая химия: Учебник для вузов: В 2 т / В. Ф. Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — С. 237—305. — ISBN 5-94628-171-2.
Механизмы реакций с участием алкенов
- Марч Дж. Глава 15. Реакции присоединения к кратным связям углерод-углерод. Глава 16. Реакции присоединения к кратным связям углерод-гетероатом. // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах / Пер. с англ., под редакцией И. П. Белецкой. — М.: Мир, 1988. — Т. 3. — С. 132—430.
- Сайкс П. Механизмы реакций в органической химии / Пер. с англ.,под редакцией В. Ф. Травеня. — 4-е изд. — М.: Химия, 1991. — 448 с. — ISBN 5-7245-0191-0.
Использование алкенов в промышленности
Примечания
- Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — 727 с. — ISBN 5-94628-171-2.
- Мазалов Л.Н. Электронно-структурные факторы в экстракции (недоступная ссылка). Журнал структурной химии. ИНХ СО РАН (17 октября 2002). Дата обращения: 24 июля 2009. Архивировано 20 сентября 2008 года.
- Случайные открытия. Этилен (недоступная ссылка). Занимательная химия. Дата обращения: 22 июля 2009. Архивировано 19 мая 2007 года.
- Открытие этилена (pdf). Открытия в органической химии и биохимии. Единая коллекция цифровых образовательных ресурсов. Дата обращения: 22 июля 2009.
- Меншуткин Н. Очеркъ развитія химическихъ воззрҌній. — С-Петербургъ: Тип. В.Демакова, 1888. — С. 252—264.
- Фигуровский Н.А. История химии: Учеб. пособие для студентов пед. ин-тов по хим. и биол. спец. — М.: Просвещение, 1979. — С. 102.
- Соловьев Ю. И. История химии: Развитие химии с древнейших времен до конца XIX в. Пособие для учителей. — 2-е изд., перераб. — М.: Просвещение, 1983. — С. 208.
- В природе существует большое количество соединений с двойными связями, например терпены или каротиноиды, однако их относят к отдельным классам соединений и в настоящей статье они не рассматриваются.
- Вредные вещества. Непредельные углеводороды этиленового ряда (алкены). Новый справочник химика и технолога. Chemanalytica.com. Дата обращения: 22 июля 2009.
- Непредельные, или ненасыщенные, углеводороды ряда этилена (алкены). Органическая химия. Chemistry.narod.ru. Дата обращения: 22 июля 2009. Архивировано 28 мая 2013 года.
- Свойства органических соединений. Справочник. / Под. ред. А. А. Потехина. Л. Химия. — 1984. — 520 с.
- The Nobel Prize in Chemistry 1979 (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation. Дата обращения: 25 июля 2009. Архивировано 22 августа 2011 года.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 3. — 459 с.
- Робертс Дж., Касерио М. Основы органической химии = Basic principles of organic chemistry / Под редакцией академика Несмеянова А.Н.. — 2-е, дополненное. — М.: Мир, 1978. — Т. 1. — С. 227—228.
- Кондакова реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 887—888.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 2. — 504 с.
- Курц А Л., Ливанцов М.В., Ливанцова Л.И. Карбены и карбеноиды (раздел 4.7.). Алкены (часть II). Химический факультет МГУ. Дата обращения: 22 июля 2009.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 1. — С. 253.
- Ли Дж. Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — 456 с. — ISBN 5-94774-368-X.
- Курц А Л., Ливанцов М.В., Ливанцова Л.И. Химические свойства алкенов (раздел 4.). Алкены (часть II). Химический факультет МГУ. Дата обращения: 22 июля 2009.
- Макквиллин Ф. Дж. Гомогенное гидрирование в органической химии = Homogeneous hydrogenation in organic chemistry / Пер. с англ. Н.М.Лойма. — М.: Химия, 1980. — 160 с.
- Воля-Циглера реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 824—825.
- Хейнс А. Методы окисления органических соединений: Алканы, алкены, алкины и арены = Methods for the oxidation of organic compounds: Alkanes, Alkenes, Alkynes and Arenes / Перевод с англ., под редакцией И.П. Белецкой. — М.: Мир, 1988. — 400 с. — ISBN 5-03-000149-2.
- Прилежаева реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 4. — С. 169.
- Фальбе Ю. Синтез на основе окиси углерода / Пер. с нем. — Л., 1971.
- The Nobel Prize in Chemistry 2005 (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation. Дата обращения: 22 июля 2009. Архивировано 22 августа 2011 года.
- Механизм реакции метатезиса (jpg) (недоступная ссылка — история ). Сайт журнала "Наука и жизнь". Дата обращения: 22 июля 2009. (недоступная ссылка)
- Нобелевскую премию по химии присудили Иву Шавену, Роберту Груббсу и Ричарду Шроку. Новости. Lenta.ru (5 октября 2005). Дата обращения: 22 июля 2009.
- Метатезис в водной среде. Новости химической науки. Портал Chemport.ru (9 февраля 2008). Дата обращения: 22 июля 2009.
- Олефины // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 3. — С. 737—740.
- Дегидрирование алканов (раздел 2.5.3.) (недоступная ссылка). Интерактивный мультимедиа учебник "Органическая химия". Самарский ГУ, Кафедра органической, биорганической и медицинской химии. Дата обращения: 22 июля 2009. Архивировано 28 октября 2011 года.
- Алкены и алкадиены из алканов (недоступная ссылка). Нефтехимия. Chemistry.narod.ru. Дата обращения: 22 июля 2009. Архивировано 16 октября 2009 года.
- Катализаторы дегидрирования // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 670—671.
- Нейланд О. Я. Органическая химия: Учеб. для хим. вузов. — М.: «Высшая школа», 1990. — 750 с. — ISBN 5-06-001471-1.
- Матьё Ж., Панико Р., Вейль-Рейналь Ж. Изменение и введение функций в органическом синтезе = L'amenagement fonctionnel en synthese organique / Перевод с французского С.С. Юфита. — М.: «Мир», 1980. — С. 169.
- Керри Ф, Сандберг Р. Книга первая. Структура и механизмы // Углубленный курс органической химии / Пер. с англ., под редакцией проф. В.М.Потапова. — М.: Химия, 1981. — С. 54—59.
- Хорнера реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 5. — С. 606—607.
- Реакция Чугаева (недоступная ссылка). Именные органические реакции. Иркутский государственный университет. Химический факультет. Дата обращения: 22 июля 2009. Архивировано 19 апреля 2011 года.
- Аммониевые соединения // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 278—280.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 4. — С. 49—53.
- Реакция Бурда (недоступная ссылка). Именные органические реакции. Иркутский государственный университет. Химический факультет. Дата обращения: 22 июля 2009. Архивировано 10 апреля 2013 года.
- Бэмфорда-Стивенса реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 658.
- Дядченко В.П., Андресюк А.Н, Белоглазкина Е.К., Брусова Г.П. Использование защитных групп в синтезе. Планирование многостадийных синтезов. Химический факультет МГУ (2003). Дата обращения: 25 июля 2009.
- Перкина реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 3. — С. 965—966.
- Шрайнер Р., Фьюзон Р., Кёртин Д., Моррилл Т. Идентификация органических соединений. Химический каталог. Дата обращения: 22 июля 2009.
- Вульфсон Н.С., Заикин В.Г., Микая А.И. Масс-спектрометрия органических соединений. — Химия. — М., 1986. — С. 31.
- Микая А.И., Сметанин В.И., Заикин В.Г. // Серия химическая : Сб. — Известия АН СССР, 1982. — С. 2214.
- Казицына Л.А., Куплетская Н.Б. Применение УФ-, ИК- и ЯМР-спектроскопии в органической химии. — М.: Высшая школа, 1971. — С. 66—67.
- Браун Д., Флойд А., Сейнзбери М. Спектроскопия органических веществ = Organic Spectroscopy / Пер. с англ. А. А. Кирюшкина. — М.: Мир, 1992. — С. 50. — ISBN 5-03-002111-6.
- Ионин Б.И., Ершов Б.А., Кольцов А.И. ЯМР-спектроскопия в органической химии / Под ред. Ершова Б.А.. — 2-е изд., перераб. — Л.: Химия, 1983. — С. 157—158.
- Прогноз рынка этилена. Конъюнктура. Товары и рынки. Российский Центр внешней торговли. Дата обращения: 22 июля 2009.
- Этилен, этен. Статьи о газах. Компания "НИИ КМ". Дата обращения: 22 июля 2009.
- Мировой рынок пропилена. Ssa.ru. Дата обращения: 22 июля 2009.
- Метатезис олефинов: современный путь к полипропилену. Аналитический портал химической промышленности: Новые химические технологии. Дата обращения: 22 июля 2009.
- Бутены // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 638—640.
| Углеводороды | |
|---|---|
| Кислородсодержащие | |
| Азотсодержащие | |
| Серосодержащие | |
| Фосфорсодержащие | |
| Галогенорганические | |
| Кремнийорганические | |
| Элементоорганические | |
| Другие важные классы | |

{kind=link}
{kind=link}