Гидрирование
Гидри́рование (гидрогениза́ция) — химическая реакция, включающая присоединение водорода к органическому веществу. В ходе данной реакции молекула водорода присоединяется к двойной или тройной связи молекулы. Если в результате гидрирования происходит разрыв связи углерод — углерод или углерод — гетероатом, то такой процесс называется гидрогенолизом[1].
Гидрирование широко применяется для получения органических веществ как в лаборатории, так и в промышленном масштабе. Оно также используется в некоторых процессах очистки, например, для удаления следов ацетилена из этилена или примесей кислорода из различных систем[1].
История
Первая описанная в литературе реакция каталитического гидрирования была проведена в 1874 году. Она заключалась в превращении ацетилена и этилена в этан. Расцвет каталитического гидрирования был связан с работами Поля Сабатье, который превратил гидрирование в универсальный метод и в 1912 году получил за это Нобелевскую премию. В оригинальной работе водород и пары органического вещества пропускали над медным или никелевым катализатором при температурах 100—300 °C. В данное время такой метод гидрирования не используется[2].
Механизм
Гидрирование находится в равновесии с обратным процессом дегидрирования и является сильно экзотермическим процессом (105—125 кДж на 1 моль водорода). Согласно принципу Ле Шателье, такое равновесие должно смещаться вправо при понижении температуры, поэтому в промышленных реакциях иногда ограничивают температуру, повышающуюся из-за экзотермичности реакции[3].

Гетерогенный катализ
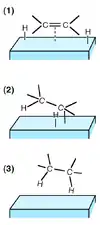
За небольшими исключениями, молекулярный водород не реагирует с органическими веществами при температуре ниже 480 °С. Реакция с газообразным водородом возможна лишь на поверхности катализатора, который сорбирует и водород, и органические молекулы, облегчая их контакт. Однако даже в таких условиях энергия активации реакции составляет 6,5—16 ккал/моль (значения измерены для реакции пропилена с водородом в присутствии различных катализаторов). Активность металлических катализаторов в данной реакции уменьшается в следующем ряду[4]:

Из металлов данного ряда сейчас используются только платина, палладий, родий, рутений и никель. Также находят применение и некоторые другие вещества[4].
Согласно механизму Хориути — Поляни (1934), реакция на поверхности катализатора осуществляется постадийно. На первой стадии происходит сорбция субстрата и водорода на катализаторе, затем на второй стадии атом водорода мигрирует к β-углеродному атому двойной связи и образуется связь между металлом и α-углеродным атомом. Наконец, на третьей стадии происходит восстановительное элиминирование продукта. Между присоединением первого и второго атома водорода проходит некоторое время, за которое могут происходить водородный обмен, цис-транс-изомеризация или миграция двойной связи. Побочные процессы также ускоряются при недостаточном насыщении катализатора водородом. Гидрирование протекает тем быстрее, чем менее затруднена молекула. Скорость гидрирования алкенов уменьшается при увеличении числа заместителей при двойной связи. Электронные эффекты гораздо меньше влияют на реакцию[4][5].
Считается, что атомы водорода, находящиеся на поверхности катализатора, присоединяются к молекуле субстрата с одной стороны, из-за чего образуются продукты цис-конфигурации[5].
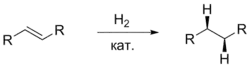
Гомогенный катализ
При гомогенном катализе водород и гидрируемое вещество координируются внутри каталитического комплекса. При этом водород диссоциирует, за счёт чего и происходит его активация[6]. Активация водорода может происходить тремя способами, из которых лишь третий важен для рассмотрения механизма гомогенного катализа реакции гидрирования:
- путём окислительного присоединения;
- путём гомолиза;
- путём гетеролиза[7].
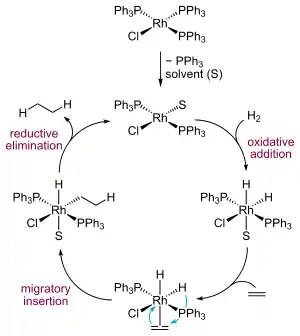
Активация алкена происходит также за счёт координации с атомом металла. При этом алкены никогда не вытесняют из комплекса другие лиганды, поэтому их координация может происходить только к ненасыщенным комплексам, которые должны образовываться в реакционной смеси. Способность алкена связываться с металлом во многом зависит от доступности его двойной связи и её конфигурации. Образующиеся алкеновые комплексы переходных металлов не очень активно активируют молекулярный водород, поэтому большинство известных каталитических систем реагируют по дигидридному каталитическому циклу. (Описаны также алкеновый и алкильный цикл, однако они встречаются редко.) Типичным примером такого поведения служит катализатор Уилкинсона, который реагирует с водородом, образовывая дигидридный комплекс. Далее происходит координация с алкеном, его миграция и восстановительное элиминирование продукта, после чего ключевой каталитический интермедиат вовлекается в очередной цикл реакции[7].
Лимитирующей стадией является атака алкена на дигидридный комплекс. Недиссоциированные комплексы родия с этиленом не могут активировать кислород, поэтому этилен отравляет катализатор в своей собственной реакции восстановления и является нежелательной примесью при гомогенном восстановлении других алкенов[8].
Несмотря на то, что присоединение атомов водорода происходит не одновременно, стереохимический результат можно описать как син-присоединение (то есть с образованием цис-продуктов). Причина такой стереоселективности в том, что родий и водород присоединяются к двум углеродным атомам двойной связи синхронно. Второй атом водорода внедряется по связи углерод — родий, сохраняя общую цис-конфигурацию. Показано также, что водород преимущественно присоединяется с более доступной стороны двойной связи, что затем было использовано при дизайне хиральных катализаторов гомогенного гидрирования[8].
Гомогенный катализ дигидридного типа имеет ряд преимуществ перед гетерогенным катализом и гомогенным катализом других типов. Короткое время жизни алкильного комплекса уменьшает вероятность реакций изомеризации. Это свойство также позволяет проводить реакции дейтерирования[8].
Условия реакции
Каталитическое гидрирование развилось в два основных направления: гидрирование в стеклянном приборе при низких температурах (до 100 °C) и низком давлении (1—4 атм) и гидрирование при высоком давлении (от нескольких до нескольких сотен атм) и температурах от 20 до 400 °С. Второй метод гидрирования требует более сложных приборов — автоклавов, выдерживающих давление до 350 атм. В лабораторных условиях гидрирование под высоким давлением проводят в небольших стальных цилиндрах, присоединённых к источнику водорода и насосу, перемешивание в которых осуществляется при помощи магнитной мешалки, а нагревание — на масляной бане[2].
Субстраты
Гидрирование может применяться для соединений различных классов. Используемые субстраты и соответствующие им продукты приведены в таблице[3].
| Субстрат | Формула субстрата | Продукт | Формула продукта |
|---|---|---|---|
| алкены | R2C=CR2 | алканы | R2CH-CHR2 |
| алкины | RC≡CR | алкены | цис-RCH=CHR |
| альдегиды | RCHO | первичные спирты | RCH2OH |
| кетоны | RCOR' | вторичные спирты | RR’CHOH |
| карбоновые кислоты | RCOOH | первичные спирты | RCH2OH |
| сложные эфиры | RCOOR' | два спирта | RCH2OH, R’OH |
| имины | R2C=NR' | амины | R2CH-NHR' |
| амиды | RC(O)NR'2 | амины | RCH2NR'2 |
| нитрилы | RCN | имины | RCH=NH |
| нитросоединения | RNO2 | амины | RNH2 |
| сульфиды | RSR' | насыщенные соединения | RH, R’H, H2S |
Катализаторы
Для проведения реакции гидрирования используют широкий набор катализаторов. Достаточно активными являются металлы группы платины: платина, палладий, родий и рутений. В качестве экономичных альтернатив предложены недрагоценные металлы: никель (Алюминат никеля(II)), медь, молибден и кобальт. Данные металлы обладают способностью сорбировать одновременно субстрат и водород, облегчая реакцию между ними[9].
Катализаторы гидрирования делятся на две группы:
- гетерогенные катализаторы, являющиеся твёрдыми веществами и образующие отдельную фазу в жидкой реакционной смеси;
- гомогенные катализаторы, растворяющиеся в жидкой среде[9].
Гетерогенные катализаторы
- Платиновые катализаторы
Первоначально предложенные виды платины (коллоидная платина, платиновая губка) вышли из употребления: на смену им пришли катализаторы с более воспроизводимыми свойствами. Оксид платины PtO2 (катализатор Адамса) представляет собой стабильный коричневый порошок, который под действием водорода превращается в платину с очень высокой активностью. Он подходит почти для всех реакций гидрирования, активируется некоторыми солями металлов, дезактивируется серой и другими каталитическими ядами, выдерживает сильные органические и минеральные кислоты[10].
Для увеличения площади контакта катализатора с водородом и гидрируемыми веществами платину осаждают на специальные подложки (активированный уголь, силикагель, сульфат бария и другие). Это достигается путём восстановления платинохлористоводородной кислоты в водных суспензиях данных материалов. Такие катализаторы содержат 5, 10 или 30 % платины по массе, имеют высокую активность, часто проявляют пирофорность[10].
Платиновые катализаторы могут служить для гидрирования разнообразных веществ при комнатной температуре и невысоком давлении (1—4 атм), но они неэффективны при восстановлении карбоновых кислот или сложных эфиров до спиртов[10].
- Палладиевые катализаторы
Палладиевые катализаторы весьма похожи на платиновые. Оксид палладия PdO получают из хлорида палладия и нитрата натрия. Металлический палладий получается при восстановлении хлорида палладия боргидридом натрия. Палладиевые катализаторы на подложках (угле, карбонате кальция, сульфате бария) содержат 5 или 10 % палладия. Катализаторы на основе палладия более часто модифицируют для получения заданной селективности. Например, палладий на карбонате кальция, дезактивированный ацетатом свинца, служит для частичного гидрирования алкинов до цис-алкенов (катализатор Линдлара)[10].
Палладиевые катализаторы можно использовать в сильнокислой и щелочной среде. Они подходят для гидрогенолиза защитных групп бензильного типа[10].
- Никелевые катализаторы
Катализаторы на основе никеля универсальны и используются как в лаборатории, так и в промышленности. В качестве носителя используется кизельгур, в водной суспензии которого осаждают карбонат никеля (действием карбоната натрия на нитрат никеля), который затем восстанавливают водородом при 450 °C, после чего сушат при 110—120 °С[10].
Весьма активными являются катализаторы типа никеля Ренея: их получают из сплава никеля и алюминия путём растворения в 25—50 % растворе гидроксида натрия при нагревании. Алюминий растворяется, а никель остаётся в виде очень мелкого порошка. В зависимости от условий образуется катализатор той или иной активности. Никель Ренея может использоваться для восстановления практически любых функциональных групп, он не дезактивируется серой и может быть использован для десульфурирования серосодержащих соединений. Катализаторы P-1 и P-2, похожие по активности на никель Ренея, получают восстановлением солей никеля, например, ацетата никеля, боргидридом натрия. Они содержат большую долю борида никеля, не пирофорны и могут применяться для гидрирования при комнатной температуре и атмосферном давлении. Никель, осаждённый из раствора хлорида никеля алюминиевой или цинковой пылью, называется катализатором Урушибары и по активности также похож на никель Ренея[10].
- Другие гетерогенные катализаторы
В реакциях гидрирования применяются также катализаторы на основе оксидов меди, цинка и хрома, однако их использование ограниченно, поскольку они требуют высоких температур (150—200° C) и давления (100—150 атм). То же самое касается рениевых катализаторов[10].
Гомогенные катализаторы
Отдельный класс катализаторов гидрирования составляют соединения, растворимые в органических растворителях, — катализаторы гомогенного гидрирования. Они представляют собой комплексы благородных металлов. Наиболее известным примером является хлорид трис(трифенилфосфин)родия. Гомогенное гидрирование обычно проводят при комнатной температуре и атмосферном давлении. Оно менее эффективно и более селективно, чем гетерогенное гидрирование, поэтому больше подходит для восстановления сложных полифункциональных субстратов. Также данные катализаторы применяются в энантиоселективном гидрировании, поскольку предоставляют большие возможности для введения хиральных лигандов[10].
В промышленности такие катализаторы используются только в тех случаях, когда не удалось подобрать подходящий гетерогенный катализатор. Это связано с тем, что их трудно выделить из реакционной смеси. Тем не менее, хиральный катализатор на основе родия используется в промышленном синтезе леводопы[11].
Активность катализаторов
На активность катализаторов большое влияние оказывает присутствие посторонних примесей, которые могут увеличить, уменьшить скорость гидрирования или даже полностью его остановить. Например, всего 0,2 % примеси палладия на платиновом катализаторе с угольной подложкой деактивирует этот катализатор в реакции гидрогенолиза бензильных защитных групп и галогенов[10].
Самыми сильными ингибиторами благородных металлов являются сера и большинство серосодержащих соединений. За исключением отдельных случаев (например, использования катализатора Линдлара) их присутствие в реакционной смеси весьма нежелательно. Удаление их из среды можно провести под действием никеля Ренея, который связывает серу в виде сульфида никеля. Встряхивание или перемешивание реакционной смеси с этим катализатором делает возможным дальнейшее гидрирование в присутствии благородных металлов. Многие нуклеофилы (меркаптаны, сульфиды, цианиды, иодиды) выступают как ингибиторы по отношению к платине, палладию и родию[10].
Большую роль играет также кислотность среды: для гидрирования ароматических колец необходимо наличие кислоты, а реакции с участием никеля Ренея обычно проводят в присутствии третичных аминов или щелочей[10].
Другие условия
Каталитическое гидрирование проводят в присутствии 1—3 % платинового катализатора (в расчёте на массу металла). Никель Ренея используется в гораздо бо́льших количествах. Лучшие растворители для водорода (пентан, гексан) не всегда являются таковыми для остальных компонентов реакции. Растворяющая способность метанола и этанола по отношению к водороду в три раза ниже, однако они используются наиболее часто. Также в качестве растворителей для проведения гидрирования используют бензол, циклогексан, диоксан и уксусную кислоту. Вода также может быть использована для гидрирования тех веществ, которые в ней растворимы (например, кислот и их солей)[12].
На скорость реакции влияет температура, хоть это влияние не так сильно, как в случае других реакций. Повышение давления ожидаемо увеличивает скорость гидрирования. Поскольку реакция является трёхфазной, эффективным перемешиванием также не следует пренебрегать[12].
Проведение реакции
Для проведения реакции необходимо рассчитать количество водорода, которое необходимо затратить на гидрирование субстрата. Особенно это важно для реакций частичного гидрирования, когда требуется вовремя остановить реакцию. В точных расчётах необходимо учитывать давление пара растворителя, поскольку оно также вносит вклад в общее давление в системе. Если в качестве катализатора используются оксиды металлов, необходимо также учесть количество водорода, необходимое для их восстановления[13].
При смешивании реагентов следует аккуратно обращаться с пирофорными катализаторами: обычно раствор добавляют к катализатору, находящемуся в реакционном сосуде, а не наоборот[13].
Выделение продукта проводят фильтрованием катализатора и упариванием растворителя. Перегонка или перекристаллизация обычно приводят к чистому продукту. Если используется гомогенный катализатор, то обработка реакционной смеси более сложная и зависит от типа катализатора[13].
Стереоселективное гидрирование
Родиевый асимметрический катализ
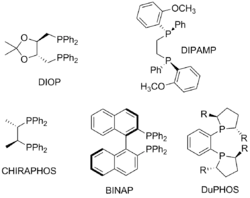
Первые примеры асимметрической реакции гидрирования, катализируемой комплексом родия, были опубликованы независимо Хорнером и Ноулзом в 1968 году (Нобелевская премия, 2001). Катализатор Уилкинсона, содержащий в качестве лиганда хиральный фосфин P(C6H5)(н-С3H7)(CH3) катализировал гидрирование некоторых алкенов с оптическим выходом в 3—15 %. Прогресс в асимметрическом гидрировании начался с открытием бидентатных хиральных фосфиновых лигандов. Так, Анри Каган открыл лиганд DIOP, получаемый из винной кислоты. Родиевый комплекс с этим лигандом катализировал энантиоселективное гидрирование производных α-(ациламино)коричной кислоты до соответствующих аминокислот с энантиомерным избытком до 80 %. Позже был найден другой отличный лиганд для этой реакции — DIPAMP. Поиск новых лигандов позволил наладить промышленное получения ряда природных и неприродных аминокислот с энантиомерным избытком более 90 %[14].

При изучении области применимости данной реакции было обнаружено, что она не очень широка: необходимо, чтобы при двойной связи находилась аминогруппа и атом водорода в транс-положении, иначе невозможно добиться высокой энантиоселективности. Фенильная группа может быть заменена водородом либо углеводородным заместителем[14].
Рутениевый асимметрический катализ
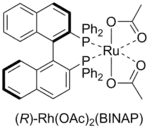
Вместе с тем разрабатывались методы энантиоселективного гидрирования, заключающиеся в использовании рутениевых катализаторов (Р. Ноёри, Нобелевская премия, 2001). В частности, гидрирование α,β-ненасыщенных карбоновых кислот протекало с количественным выходом и высокой энантиоселективностью при использовании хирального фосфин-содержащего катализатора Ru(OAc)2(BINAP), при этом необходимое количество используемого катализатора было меньше количества субстрата в 100—600 раз. На основе данной реакции был разработан промышленный энантиоселективный синтез противовоспалительного препарата напроксена[15].
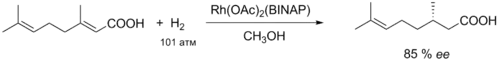
Подобным образом енамиды, содержащие при двойной связи донорную группировку, также подвергаются гидрированию в присутствии хиральных рутениевых катализаторов с высокой энантиоселективностью. Впоследствии такие процессы были развиты в общий асимметрический синтез алкалоидов изохинолинового ряда. Рутениевый катализ был применён также к ряду других субстратов, например, для тех же α-(ациламино)коричных кислот, гидрирование которых разрабатывалось У. Ноулзом. Интересно, что направление асимметрической индукции в данном случае оказалось противоположным тому, что наблюдалось в случае родиевого катализа. В реакцию эннатиоселективного гидрирования вступали также β-(ациламино)акриловые кислоты, аллиловые и гомоаллиловые спирты[16].
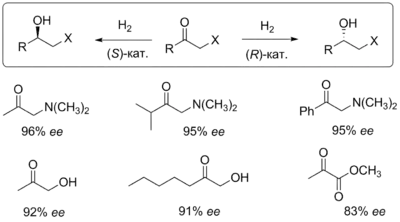
Стереоселективное гидрирование кетонов
Гидрирование кетонов было реализовано с использованием хиральных родиевых катализаторов и дало возможность энантиоселективно синтезировать адреналин, пантолактон и ряд бета-адреноблокаторов. Рутениевый катализ также позволяет гидрировать карбонильные соединения. Реакция требует наличия рядом с карбонильной группой какой-либо стереонаправляющей функциональной группы в α-, β- или γ-положении (диалкиламино, гидроксильной, алкоксильной, кетогруппы, сложноэфирной, карбоксильной или др.), а направление асимметрической индукции можно изменять путём смены конфигурации лиганда BINAP в рутениевом катализаторе. Наилучшими субстратами являются β-кетоэфиры. Например, реакция гидрирования метил-3-оксобутаноата даёт выход, близкий к количественному, практически 100%-ный оптический выход и позволяет проводить синтез в масштабе от 100 мг до 100 кг при весьма низкой концентрации катализатора (отношение концентраций субстрата и катализатора составляет от 1000 до 10 000)[17].
Гидрирование жиров
Природные жиры и масла обладают уникальными физико-химическими и пищевыми свойствами, определяемыми их триглицеридным составом. В твёрдых жирах обычно высока доля насыщенных жирных кислот, в то время как жидкие масла богаты моно- и полиненасыщенными кислотами. Тем не менее, жиры в своём природном виде не всегда подходят для применения в промышленности[18][19].
Гидрогенизация (гидрирование), наряду с фракционированием и переэтерификацией, является традиционным хорошо изученным промышленным процессом модификации жиров. Процесс был открыт в начале XX в. и стал революционным для масложировой промышленности, поскольку позволял из жидких масел получать твёрдые и полутвёрдые жиры с нужной консистенцией. С химической точки зрения гидрогенизация представляет собой реакцию присоединения газообразного водорода к ненасыщенным (двойным или тройным) связям в молекулах жиров в присутствии катализатора. В результате получается твёрдый или полутвёрдый жир с высокой устойчивостью к окислению[20].
История гидрирования жиров
До XX в. основным жировым продуктом, наиболее используемым как производителями, так и рядовыми потребителями, являлось сливочное масло. Благодаря высокому содержанию насыщенных кислот масло обладает необходимой твёрдостью для использования в выпечке, слоёных изделиях, кремах; то же время оно достаточно пластичное, чтобы его можно было намазать на хлеб.
Производство заменителей сливочного масла из-за его высокой цены началось в Европе в середине XIX в. По приказу Наполеона Бонапарта французский химик Ипполит Меж-Мурье получил такой подходящий заменитель. Данный продукт был получен из фракции говяжьего жира и обладал отличной консистенцией и плавлением во рту, что пришлось по нраву потребителям. Впоследствии для производства заменителей использовался также свиной жир[21].
В 1897 г. французские химики Поль Сабатье и Жан-Батист Сандеран разработали процесс присоединения газообразного водорода к жиру в присутствии катализатора – гидрирование, позволяющее получать твёрдые жиры из жидких растительных масел и жиров морских животных[22].
Гидрогенизации нашли промышленное применение в Англии в 1903 г., когда стали получать твёрдый жир из жира кашалотов. В 1909 г. процесс использовали для получения заменителей животных жиров ввиду нехватки последних для мыловаренной промышленности. В дальнейшем жиры и маргарины из гидрогенизированных хлопкового и соевого масел стали получать в Европе и США[23][24].
Начиная с 1930-х гг. промышленная гидрогенизация развивается огромными темпами вследствие колоссального потребления маргаринов и гидрированных жиров в ходе Второй мировой войны. Тем не менее, до 1940 г. маргарин рассматривался как низкокачественный заменитель сливочного масла. В 1941 г. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) присвоило маргаринам статус основного продовольственного продукта[21][24].
Цели и процесс гидрирования жиров
В общем случае, гидрогенизация жиров преследует следующие цели[25]:
1. Превратить жидкое масло в твёрдый жир. Когда природные твёрдые жиры требуемой консистенции слишком дорогие, гидрогенизация (зачастую в комбинации с переэтерификацией и/или фракционированием) позволяет получить жир с желаемыми свойствами.
2. Изменить консистенцию жира. Температуру плавления гидрогенизированного жира можно контролировать степенью гидрирования. Кроме того, цис-изомеры, содержащиеся в растительных маслах, в ходе гидрогенизации превращаются в транс-изомеры, которые придают жиру другие свойства плавления.
3. Стабилизировать жир. Поскольку насыщенные кислоты менее подвержены окислению по сравнению с ненасыщенными, а транс-изомеры – по сравнению с цис-изомерами, гидрированные жиры обладают более высокой окислительной стабильностью и большим сроком годности по сравнению с жидкими маслами.
В ходе гидрогенизации проходит два основных процесса: присоединение водорода к ненасыщенным связям в молекулах жиров, т.е. непосредственно гидрирование; изомеризация части ненасыщенных связей с образованием транс-изомеров жирных кислот[26].
Гидрогенизация преимущественно зависит от температуры и длительности реакции, давления водорода, скорости перемешивания, типа и концентрации катализатора.
В зависимости от глубины протекания реакции гидрогенизация подразделяется на:
- частичную, в ходе которой только часть ненасыщенных связей в жире взаимодействует с водородом. В частично гидрогенизированном жире сохраняется определённая доля ненасыщенных связей, а содержание транс-изомеров жирных кислот может составлять от 20 до 60 %[27].
- полную, в ходе которой все ненасыщенные связи в жире взаимодействуют с водородом. Поскольку в этом случае не остаётся ненасыщенных связей, которые могут подвергнуться изомеризации, полностью гидрогенизированные жиры не содержат транс-изомеров[28].
Катализаторы гидрирования жиров
Наиболее изученными катализаторами гидрирования жиров являются медь, никель, палладий и платина[29].
Катализаторы на основе меди обладают высокой селективностью (избирательностью) по отношению к α-линоленовой и линолевой кислотам с малой склонностью к образованию транс-изомеров. В 1970 – 1980 гг. эти катализаторы широко использовались для частичной гидрогенизации. Тем не менее, из-за низкой активности и усиления окисления ненасыщенных жирных кислот их использование сократилось[30].
Катализаторы на основе благородных металлов (палладия и платины) обладают высокой селективностью, позволяя проводить гидрирование с той же скоростью при меньшей температуре по сравнению с никелем[23]. Несмотря на то что такие условия снижают образование транс-изомеров, повышенная каталитическая активность приводит к снижению дозировок катализатора, что требует повышения эффективности фильтрации и потому ограничивает их применение[29].
В результате на практике в качестве катализатора используется преимущественно никель вследствие его достаточно высокой активности, селективности, лёгкости фильтрации от жира, возможности повторного использования, малого влияния на окисление ненасыщенных кислот и относительно невысокой стоимости (по сравнению с платиной и палладием)[29][31].
Современное состояние
С 1990-х годов появилось множество публикаций, указывающих на увеличение риска сердечно-сосудистых заболеваний (ССЗ) от потребления транс-изомеров жирных кислот, что спровоцировало дебаты вокруг этой проблемы в академических кругах. Исследования 1980 – 1990-х годов подтвердили связь потребления транс-изомеров жирных кислот с концентрацией липопротеинов низкой плотности («плохого холестерина») в крови и риском ишемической болезни коронарных сосудов сердца[32][33][34]. Всемирная организация здравоохранения рекомендует населению свести к нулю потребление промышленных транс-жиров[35].
Медицинские исследования и, как следствие, негативное отношение потребителей к транс-жирам и их законодательное ограничение привело к тому, что с начала XXI в. пищевая промышленность поэтапно отказывается от использования частично гидрогенизированных жиров в сторону полностью гидрогенизированных и переэтерифицированных жиров. В Дании запрет на промышленные транс-жиры введён с 2003 г.[36] В США Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) в 2015 г. постановило, что транс-жиры «не могут более добавляться в пищевые продукты после 18 июня 2018 г., если производителем не представлены убедительные научные доказательства, что их использование не представляет опасности»[37][38].
В России содержание транс-изомеров жирных кислот в масложировой продукции регулируется Техническим регламентом Таможенного союза № 024/2011 «На масложировую продукцию» и с 1 января 2018 г. составляет не более 2 % для эквивалентов масла какао, улучшителей масла какао SOS-типа и заменителей масла какао POP-типа, маргаринов, заменителей молочного жира и жиров специального назначения[39].
Применение
Наиболее важными примерами гидрирования в промышленности являются синтез Фишера — Тропша (гидрирование оксида углерода(II) для получения метанола и углеводородов), гидрирование угля, использовавшееся во времена Второй мировой войны для получения жидкого топлива, а также гидрирование азота с образованием аммиака (процесс Габера — Боша)[1].
Примечания
- Ullmann, 2012, p. 451.
- Hudlicky, 1984, p. 1—4.
- Ullmann, 2012, p. 452.
- Hudlicky, 1984, p. 4—5.
- Encyclopedia of Catalysis, 2002.
- Кнунянц И. Л. и др. Т. 1 А—Дарзана // Химическая энциклопедия. — М.: Советская энциклопедия, 1988. — С. 553—554. — 100 000 экз.
- The Metal – Carbon Bond, 1987, p. 1053—1055.
- The Metal – Carbon Bond, 1987, p. 1055—1061.
- Ullmann, 2012, p. 452—453.
- Hudlicky, 1984, p. 5—11.
- Ullmann, 2012, p. 453.
- Hudlicky, 1984, p. 11—12.
- Hudlicky, 1984, p. 12—13.
- Noyori, 1994, p. 17—28.
- Noyori, 1994, p. 28—33.
- Noyori, 1994, p. 33—47.
- Noyori, 1994, p. 56—70.
- Sivakanthan, S.; Madhujith, T. Current trends in applications of enzymatic interesterification of fats and oils: A review. LWT 2020, 132, 109880.
- Kadhum, A.A.H.; Shamma, M.N. Edible lipids modification processes: A review. Crit. Rev. Food Sci. Nutr. 2017, 57, 48–58.
- Temkov M.; Muresan V. Tailoring the Structure of Lipids, Oleogels and Fat Replacers by Different Approaches for Solving the Trans-Fat Issue—A Review. Foods 2021, 10, 1376.
- Ghotra, B.S.; Dyal, S.D.; Narine, S.S. Lipid shortenings: a review. Food Res. Inter. 2002, 35, 1015.
- Gunstone, F.D. Movements towards tailor-made fats. Progr. Lipid Res. 1998, 37, 277–305.
- Johnson, L.A. Recovery, refining, converting, and stabilizing edible fats and oils. In: Akoh, C.C. and Min, D.B. (Eds), Food Lipids. New York: Marcel Dekker. 1998.
- Martin, C.A. et al. Trans fatty acid-forming processes in foods: a review. Ann. Brazil. Acad. Sci. 2007, 79(2), 343-350.
- Ariaansz, R.F. Hydrogenation in practice. In AOCS Lipid Library, Edible Oil Processing. URL: https://lipidlibrary.aocs.org/edible-oil-processing/hydrogenation-in-practice (дата обращения 26.07.2021).
- Allen, R.R. Principles and catalysts for hydrogenation of fats and oils. JAOCS 1978, 55, 792–795.
- Dhaka, V. et al. Trans fats-sources, health risks and alternative approach—A review. J. Food Sci. Technol. 2011, 48, 534–541.
- Gray, J.I.; Russell, L.F. Hydrogenation catalysts – their effect on selectivity. JAOCS 1979, 56, 36–56
- Gray, J.I.; Russell, L.F. Hydrogenation catalysts – their effect on selectivity. JAOCS 1979, 56, 36–56.
- Ackman, R.G.; Mag, T.K. Trans fatty acids and the potential for less in technical products. In: Sébédio, J.L. and Christie, W.W. (Eds), Trans fatty acids in human nutrition. Dundee: The oil press. 1998.
- Okonek, D.V. et al. Precious metal catalysis for fats and oils applications. In: Latin American Congress and Exhibit on Fats and Oils Processing, 6, Campinas. Proceedings. Campinas: Sociedade Brasileira de Ó leos e Gorduras, 1995, 39– 46.
- Willet, W.C. et al. Intake of trans fatty acids and risk of coronary heart disease among women». The Lancet 1993, 341 (8845), 581-585.
- Shapiro, S. Trans fatty acid and coronary disease: the debate continues. 2. Confounding and selection bias in the data. Am. J. Public Health 1995, 85, 410-412.
- Gans, K. M., Lapane, K. Trans fatty acid and coronary disease: the debate continues. 3. What should we tell consumers? Am. J. Public Health 1995, 85, 411-412.
- Uauy, R. et al. WHO Scientific Update on trans fatty acids: summary and conclusions. Eur. J. Clin. Nutr. 63, 68-75 (2009).
- Denmark, trans fat ban pioneer: lessons for other countries. WHO, 2018.
- Final determination regarding partially hydrogenated oils. A Notice by the Food and Drug Administration on 06/17/2015. Daily J. US Gov. 2015.
- Christensen, J. FDA orders food manufacturers to stop using trans fat within three years. CNN, 2015
- Технический регламент Таможенного союза 024/2011 «На масложировую продукцию». 2011, 37 с.
Литература
- Gallezot P. Hydrogenation – Heterogeneous // Encyclopedia of Catalysis. — Wiley, 2002. — doi:10.1002/0471227617.eoc114.
- Hartley F. R., Jardine F. H. 12. Mechanism of homogeneous hydrogenation // The Metal – Carbon Bond. — Wiley, 1987. — P. 1049—1071. — doi:10.1002/9780470771778.ch12.
- Hudlicky M. Reductions in Organic Chemistry. — Ellis Horwood Limited, 1984.
- Noyori R. Asymmetric Catalysis in Organic Synthesis. — Wiley, 1994. — 400 p. — ISBN 978-0-471-57267-1.
- The Handbook of Homogeneous Hydrogenation / de Vries J. G., Elsevier C. J.. — Wiley, 2007. — ISBN 9783527311613. — doi:10.1002/9783527619382.
- Sanfilippo D., Rylander P. N. Hydrogenation and Dehydrogenation // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2012. — doi:10.1002/14356007.a13_487.pub2.
Ссылки
- Ноулз У. Asymmetric Hydrogenations (Нобелевская лекция). Дата обращения: 19 августа 2013.
- Ноёри Р. Asymmetric Catalysis: Science and Opportunities (Нобелевская лекция). Дата обращения: 19 августа 2013.