Синдром Сиппла
Синдро́м Си́ппла (МЭН II, МЭН IIa) — включает медуллярную карциному щитовидной железы (C-клеточная карцинома, в основном продуцирующая тирокальцитонин), двустороннюю (билатеральную) феохромоцитому и первичный гиперпаратиреоз (гиперплазия или аденоматоз паращитовидных желез). Возможно сочетание с амилоидозом кожи или болезнью Гиршпрунга.[3]
| МЭН тип IIa (Синдром Сиппла) | |
|---|---|
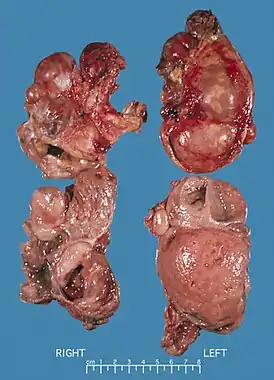 Билатеральные феохромоцитомы, ассоциированные с множественной эндокринной неоплазией типа II. Опухоль справа весом 168 г, слева - 220 г. | |
| МКБ-10 | D44.8 |
| МКБ-10-КМ | E31.22 и D44.8 |
| МКБ-9 | 258.02 |
| МКБ-9-КМ | 258.02[1][2] |
| OMIM | 171400 |
| DiseasesDB | 7984 |
| MedlinePlus | 000399 |
| eMedicine | med/1520 |
| MeSH | D018813 |
История
Впервые сочетание феохромоцитомы, медуллярной карциномы щитовидной железы и аденомы паращитовидной железы описал Сиппл в 1961 году.[4]
В 1974 году Сайзмор и соавторы (Sizemore et al) показали, что МЭН типа II объединяет две группы пациентов с феохромоцитомой и медуллярной карциномой щитовидной железы:
- МЭН IIa — с аденомой паращитовидной железы и
- МЭН IIb — без поражения паращитовидной железы, но с наличием нейрином слизистых оболочек и мезодермальными аномалиями.[5]
В 1993 ген RET-протоонкогена при МЭН типа IIa успешно клонирован.[6]
Этиология (генетические дефекты при синдроме МЭН типа IIa)
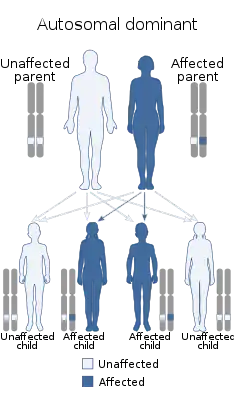
Синдром Сиппла — аутосомно-доминантно-наследуемое сочетание медуллярного рака щитовидной железы, феохромоцитомы и опухолей паращитовидных желез. Генетической основой и генетическим маркёром МЭН типа II является точечная мутация в RET-протоонкогене, локализованном в парацентромерном участке длинного плеча 10-й хромосомы, кодирующем структуру рецептора тирозинкиназы.[7] Все варианты этого синдрома обусловлены мутациями протоонкогена — у 93…95% пациентов обнаруживают точечные мутации протоонкогена c-ret (10q11), кодирующего рецептор нейротропного фактора, регулирующего пролиферацию и дифференцировку клеток, производных нервного гребня. Мутации приводят к активации рецептора c тирозинкиназной актвностью и к трансформации нейроэктодермальных клеток. Мутации могут затрагивать различные кодоны c-ret:
- у всех пациентов с классической МЭН типа IIa и МЭН типа IIa с первичным амилоидозом кожи имеются мутации в кодоне 634;
- при сопутствующей болезни Гиршпрунга возникают мутации в кодонах 634, 609, 618 и 620.[8]
Расшифровка генетических дефектов, лежащих в основе синдромов МЭН, позволяет оценивать риск развития заболевания у родственников пациентов и планировать профилактические мероприятия. При обнаружении мутации в RET-протоонкогене у родственников больных показана профилактическая экстирпация щитовидной железы.[7]
Клиническая картина
Определяется наличием соответствующего симптомокомплекса (все варианты обусловлены мутациями протоонкогена c-ret)[8]:
- классический вариант МЭН IIa — сочетание медуллярной карциномы щитовидной железы, феохромоцитомы и гиперплазии или аденомы паращитовидных желез;
- семейная изолированная медуллярная карцинома щитовидной железы — данный вариант редко включает другие опухоли;
- МЭН типа IIa с первичным амилоидозом кожи — красно-коричневые, мучительно зудящие высыпания (пятна, узелки) между лопатками или на голенях;
- МЭН типа IIa с болезнью Гиршпрунга.[8]
Медуллярная карцинома щитовидной железы
Обнаруживается практически у всех пациентов. Опухоли обычно многоочаговые, их развитию предшествует гиперплазия C-клеток.[8] Встречается в 5…18% случаев злокачественных новообразований щитовидной железы и возникает, как правило, в возрасте 15…20 лет (семейная форма) или около 45 лет (спорадическая форма). Опухоль, секретирующая тирокальцитонин, небольшого размера (до 10 мм) и весьма злокачественная — приблизительно у 40% пациентов на момент выявления обнаруживают метастазы в регионарные лимфатические узлы, которые бывают двусторонними. Метастазирует быстро:
- лимфогенным путём — в средостение;
- гематогенным путём — в лёгкие, печень человека и кости.[3]
Патофизиологическая роль усиленной секреции тирокальцитонина остаётся не выясненой. Доказано, что повышенный уровень гормона не приводит к гипокальциемии, в то же время повышенный уровень тирокальцитонина является важнейшим маркёром медуллярной карциномы щитовидной железы.[8] Часто протекает бессимптомно, иногда определяется диарея, связанная с продукцией опухолью вазоактивных веществ: серотонина, гистамина и простагландинов. Редко встречается эктопическая секреция АКТГ с клиникой гиперкортицизма.[3]
Феохромоцитома
Встречается более чем в 70% случаев, как правило, двусторонняя, многоочаговая и обнаруживается в надпочечниках или в парагнанглиях[8] (вненадпочечниковая локализация). Обычно выявляется спустя некоторое время после установления диагноза МЭН II. Редко бывает злокачественной.[3] Клиническая картина обусловлена симптоматикой феохромоцитомы — как правило, артериальная гипертония, возникающая в возрасте 20…40 лет.
J.R. Zeller и соавторы у 11 пациентов с феохромоцитомой выявили различные опухоли из островковых клеток. Эти исследователи рекомендуют обследовать всех больных с феохромоцитомой на предмет выявления опухолей из островковых клеток методом визуализации поджелудочной железы: УЗИ, сцинтиграфия, КТ или МРТ и гормонально-метаболическое исследование.[8]
Для выявления надпочечниковых и ненадпочечниковых феохромоцитом и их метастазов используют сцинтиграфию с мета-131I-бензилгуанидином или мета-123I-бензилгуанидином. Иногда выявляется и медуллярная карцинома щитовидной железы. Для сцинтиграфии также используют 99mTc-димеркаптоянтарную кислоту.[8]
Первичный гиперпаратиреоз
Встречается приблизительно в 50% случаев. Обусловлен гиперплазией или аденоматозом паращитовидных желез.[3] Вероятно, усиленная секреция тирокальцитонина не является причиной гиперпаратиреоза, поскольку он встречается и у пациентов без медуллярной карциномы щитовидной железы.[8]
Основными органами-мишенями при гиперпаратиреозе являются костная ткань, почки и желудочно-кишечный тракт. Заболевание развивается постепенно. Ранние его проявления неспецифичны: слабость, быстрая утомляемость, пациентам становится трудно ходить, ухудшается аппетит, они худеют. Присоединяется мышечная слабость, боли в ногах, походка становится «утиной». Развивается жажда, брадикардия, артериальная гипертензия. Пациенты астенизированы, вялые, депрессивны. Жалобы нарастают годами, иногда диагноз устанавливают через 10 лет с момента появления первых признаков (ранняя диагностика затруднена в связи с неспецифичностью симптоматики, а появление характе́рных проявлений позволяет распознать заболевание лишь на поздних стадиях).[3]
Первичный амилоидоз кожи
Характеризуется красно-коричневыми высыпаниями (пятна, узелки) в межлопаточной области или на голенях, которые сопровождаются сильным мучительным зудом.[3]
Болезнь Гиршпрунга
Врождённая аномалия развития — отсутствие ганглиев внутристеночных нервных сплетений на участке толстой кишки, приводящее к полному отсутствию перистальтики, расширению и атонии вышележащих участков — проявляется запорами и признаками хронической интоксикации.
Диагностика
Диагноз, как правило, выявляется случайно при скрининге узлового зоба или при обследовании родственников больного МЭН (мутация RET-протоонкогена в лейкоцитах периферической крови указывает на проявление МЭН типа II (IIa) или III (IIb); отсутствие мутации RET-протоонкогена в лейкоцитах и её наличие в опухоли — спорадическая форма медуллярной карциномы щитовидной железы).
Семейный скрининг предусматривает ежегодное исследование уровня кальцитонина в пентагастриновом тесте (стимуляция пентагастрином: 0,5 мкг/кг массы тела, забор крови на 0-й, 2-й и 5-й минуте[3]) у родственников 1-й и 2-й степени родства в возрасте от 6 до 50 лет, а также уровня экскреции катехоламинов и ванилилминдальной кислоты и кальциемии. Наиболее чувствительный генетический скрининг заключается в исследовании с целью обнаружения характерной мутации RET-протоонкогена.[7]
- УЗИ ЩЖ — обнаруживают гипоэхогенный узел в щитовидной железе, без чётких границ.
- сцинтиграфия соматостатиновых рецепторов с 99mTc-DMSA — для выявления отдалённого метастазирования медуллярной карциномы щитовидной железы.
- биопсия опухоли — находят кальцитонин, карциноэмбриональный антиген (CEA); тиреоглобулин не определяется.
Феохромоцитома при МЭН, как правило, двусторонняя (70% случаев, её клиническая картина не такая яркая, ка при спорадических формах — в плазме крови определяется повышенный уровень кальция (>2,6 ммоль/л), в моче повышена экскреция ванилил-миндальной (ВМК) и гомованилиновой кислот (ГВК), адреналина, норадреналина.
Первичный гиперпаратиреоз — в плазме крови повышены уровни кальция и паратгормона.[3]
Лечение
- Лечение начинают с удаления феохромоцитомы.
- Затем проводят экстирпацию щитовидной железы с удалением шейных лимфатических узлов и клетчатки.
- Назначают препараты тироксина для компенсации послеоперационного гипотиреоза.
- После этого приступают к лечению гиперпаратиреоза и других отклонений.
В неоперабельных случаях и если операция выполнена не радикально — назначают октреотид (синтетический аналог соматостатина) и химиотерапевтические препараты.
Первичный амилоидоз кожи и болезнь Гиршпрунга при необходимости лечатся оперативно.[3]
Прогноз
Благоприятный прогноз маловероятен.[3]
Примечания
- база данных Disease ontology (англ.) — 2016.
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- Малая энциклопедия врача-эндокринолога / Под ред. А.С. Ефимова.— К: Медкнига, 2007.— 360 с. ISBN 966-7013-23-5
- Sipple J.H. The association of pheochromocytoma with carcinoma of the thyroid gland. // Amer. J. med.— 1961,— Vol. 31.— P.163—166.
- Carney J.A. Familial multiple endocrine neoplasia: the first 100 years. // Amer. J. Surg. Pathol.— 2005,— Vol. 29, № 2, P.— 254—274.
- Donis-Keller H., Dou S., Chi D., Carlson K.M., Toshima K., Lairmore T.C., Howe J.R., Moley J.F., Goodfellow P., Wells S.A. Jr. (1993) Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. // Hum Mol Genet 2(7), P.— 851—856
- Эндокринология (краткий справочник)./Под ред. И.И. Дедова.— М., Рус. врач, 1998. — 95 с. ISBN 5-7724-0014-2
- Эндокринология. Под ред. Н. Лавина. Пер. с англ.— М., Практика, 1999. — 1128 с. ISBN 5-89816-018-3
См. также
Ссылки
| |||||||||||||||||||||||
| |||||||||||||||||||||||
| |||||||||||||||||||||||
| |||||||||||||||||||||||
Синдромы в эндокринологии | |
|---|---|
| Эпифиз |
|
| Гипоталамус |
|
| Гипофиз |
|
| Щитовидная железа |
|
| Надпочечники |
|
| Половые железы |
|
| Паращитовидные железы | |
| Островки Лангерганса | |
| Диффузная нейроэндокринная система |
|
| Прочие |
|