Синдром Нунан
Синдро́м Нуна́н (синдро́м У́льриха-Нуна́н, тернероидный синдром с нормальным кариотипом) — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом[1].
| Синдром Нунан | |
|---|---|
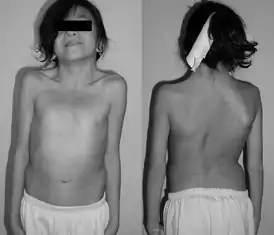 12-летняя девочка с синдромом Нунан — Типичная перепончатая шея. | |
| МКБ-11 | LD2F.15 |
| МКБ-10 | Q87.1 |
| МКБ-10-КМ | Q87.1 |
| МКБ-9 | 759.89 |
| OMIM | 163950 |
| DiseasesDB | 29094 |
| MedlinePlus | 001656 |
| eMedicine | article/947504 |
| MeSH | D009634 |
Эпидемиология
Синдром встречается у детей любого пола у 1:8 000 новорожденных (у 1:16 000 мальчиков) с нормальным кариотипом[2].
История
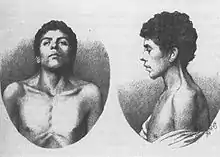
Жаклин Нунан, практиковавшая в качестве кардиолога-педиатра (детского кардиолога) в клинике университета в Айове, заметила, что дети (как мальчики, так и девочки) со стенозом лёгочной артерии (врождённый порок сердца), часто характеризуются низкорослостью, перепончатой шеей, широко расставленными глазами, птозом и низкорасположенными ушными раковинами. Изучив сочетание клинической картины врождённого порока сердца с другими аномалиями развития на примере 833 пациентов, она в 1962 году написала статью: «Associated non-cardiac malformations in children with congenital heart disease» (Сочетание несердечных аномалий у детей с врождённым пороком сердца), в которой описала 9 детей, на фоне врождённого порока сердца имевших характерные черты лица, деформации грудной клетки и невысокий рост. Патология встречалась как у мужчин, так и у женщин, при этом количество хромосом оставалось нормальным (46).
После опубликования статьи, пациентов с признаками описанными Жаклин Нунан называли «Hypertelorism with Turner Phenotype» (гипертелоризм с фенотипом Тернера). В 1971 году на симпозиуме, посвящённом вопросам кардиоваскулярной патологии для обозначения симптомокомплекса официально признан эпоним «Синдром Нунан».
Этиология и патогенез
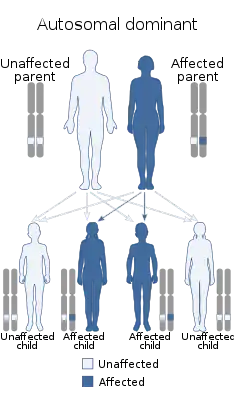
Синдром Нунан может быть вызван мутациями в различных генах, то есть это генетически гетерогенное заболевание. Большинство спорадических и семейных форм синдрома имеют аутосомно-доминантное наследования[2]. Синдром Нунан связан с мутациями в генах KRAS, NRAS, MRAS, RRAS2, SOS1, SOS2, RAF1, BRAF, RIT1, LZTR1, MAPK1, PTPN11[3].
Около 65-80% случаев синдрома Нунан связано с мутациями в генах PTPN11, SOS1 и RAF1[4].
Клиническая картина
Характеризуется низкорослостью (конечный рост у мальчиков достигает 162 см, у девочек — 153 см) и гипогонадизмом. Недостаточность гонад различна[1]:
- у мужчин — от полной агенезии до небольшой гипоплазии яичек — яички маленькие, часто отмечается крипторхизм, сперматогенез отсутствует либо выявляется различная степень олигозооспермии. Слабо развиты вторичные половые признаки (скудное половое оволосение, слабо развита мускулатура);
- у женщин часто функция яичников нормальная, могут быть менструации, возможна даже фертильность. Отмечается гипоплазия наружных гениталий.
Симптоматика[2]:
- основные клинические проявления данного синдрома имеют сходство с синдромом Шерешевского-Тернера: крыловидные складки на шее, вальгусная деформация локтевых суставов, низкорослость, лимфатические отёки кистей и стоп;
- другие проявления синдрома: птоз, впалая грудная клетка, врождённые пороки правой половины сердца (стеноз лёгочной артерии), гипертрофия левого желудочка, треугольное лицо и умственная отсталость. У мальчиков отмечаются нарушения развития яичек (крипторхизм, атрофия, анорхия, уменьшение просвета семявыносящих канальцев со склерозом или без него, уменьшение или отсутствие герминальных клеток, гиперплазия клеток Лейдига) и микропения. Некоторые пациенты с нормальными яичками сохраняют фертильность, однако у большинства отмечается умеренный или выраженный гипогонадизм. Содержание тестостерона в плазме крови низкое или определяется на нижних границах нормы, уровень гонадотропинов повышен. Кариотип XY (нормальный, мужской). Причина задержки роста не уточнена, так как уровень базального и стимулированного гормона роста нормальный. Содержание ИФР-1 снижено или остаётся на нижних границах нормы.
Диагностика
Основывается на наличии характерного фенотипа. В гормональном статусе — определяется повышенное содержание гонадотропинов в плазме крови, уровень тестостерона снижен. В эякуляте выявляется различная степень олигоспермии. Костный возраст отстаёт от паспортного. Несмотря на фенотип, характерный для синдрома Шерешевского-Тернера, при кариотипировании не находят патологического клона клеток 45, X0[1].
Лечение
При необходимости проводится заместительная терапия андрогенами. При крипторхизме показано низведение яичка[1].
Примечания
- Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 326—327. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 116-117. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
- Noonan syndrome 1. OMIM. Дата обращения: 11 января 2021.
- Bhoj E. J. et al. Phenotypic predictors and final diagnoses in patients referred for RASopathy testing by targeted next-generation sequencing (англ.) // Genetics in Medicine. — 2017. — Vol. 19, no. 6. — P. 715-718. — doi:10.1038/gim.2016.169.