Множественная эндокринная неоплазия
Мно́жественная эндокри́нная неоплази́я (МЭН) — этот термин объединяет группу наследственных аутосомно-доминантных синдромов, обусловленных опухолями или гиперплазией нескольких эндокринных желез. Возможны также смешанные типы этих синдромов[2].
| Множественная эндокринная неоплазия | |
|---|---|
| МКБ-10 | D44.8 |
| МКБ-10-КМ | D44.8 |
| МКБ-9 | 258.0 |
| МКБ-9-КМ | 258.0[1] |
| МКБ-О | 8360/1 |
| MedlinePlus | 000398 и 000399 |
| MeSH | D009377 |
Историческая справка
В 1903 году Erdheim описал у больного акромегалией случай аденомы гипофиза, сочетающийся с увеличением трёх паращитовидных желез.
В 1953 году Underdahl с соавторами описали 8 больных с сочетанием аденом гипофиза, паращитовидных желез и островковых клеток поджелудочной железы.
В 1954 году Wermer обосновал доминантный тип наследования данного симптомокомплекса.
В 1959 году Hazard с соавторами описали медуллярную (со́лидную) карциному щитовидной железы — компонент, позже включённый в состав двух эндокринных синдромов.
В 1961 году Sipple впервые опишет синдром, сочетающий феохромоцитому, медуллярную карциному щитовидной железы и аденому паращитовидных желез.
В 1966 году Williams с соавторами описали сочетание нейриномы с феохромоцитомой и медуллярной карциномой щитовидной железы.
В 1968 году Steiner с соавторами внедрили термин «множественная эндокринная неоплазия» (МЭН) для описания нарушений, обусловленных комбинацией эндокринных опухолей и предложили термины «синдром Вермера» для обозначения МЭН типа I и «синдром Сиппла» для МЭН типа II.
В 1974 году Sizemore с соавторами показали, что в состав синдрома МЭН типа II включили две категории больных с феохромоцитомой и медуллярной карциномой щитовидной железы: МЭН IIa — с аденомой паращитовидных желез и МЭН IIb — без аденомы паращитовидных желез, но с наличием нейрином слизистых оболочек и мезодермальными аномалиями. Впоследствии дополнительные неэндокринные состояния (нейрофиброматоз Реклингаузена и другие) были обнаружены в составе недавно описанных наследственных синдромов МЭН. Таким образом, сочетания нарушений, выявляемые в составе данных синдромов весьма разнообразны[3].
В 1988 году выделен локус 11-й хромосомы (11q13), ответственный за развитие синдрома Вермера (МЭН типа I).
В 1993 клонирован ген RET-протоонкогена, приводящий к развитию МЭН типа II[4].
В 1998 клонирован ген, приводящий к развитию МЭН типа I[5].
Основные черты синдромов множественной эндокринной неоплазии
- Большинство опухолей происходит из нейроэктодермы.
- Как спорадические, так и семейные случаи МЭН обусловлены генетическими дефектами и наследуются аутосомно-доминантно.
- Опухоли часто злокачественны.
- Многообразие эндокринных и метаболических нарушений:
- присутствуют симптомы, обусловленные нарушением секреции одного или нескольких гормонов;
- нередко синдромы МЭН сопровождаются дисплазией других органов и тканей[6].
Генетика
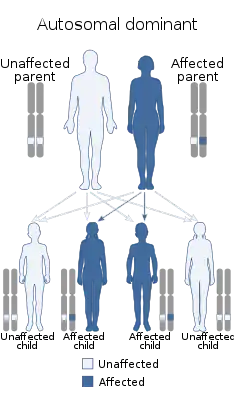
Все синдромы МЭН наследуются аутосомно-доминантно и характеризуются высокой пенетрантностью. В половине случаев множественная эндокринная неоплазия возникает спорадически, то есть обусловлена вновь появившейся мутацией в половых или соматических клетках. Генеалогические, цитогенетические и молекулярно генетические исследования выявили мутации, лежащие в основе известных типов МЭН. В семьях с МЭН риск превышает 75%[6].
Классификация
Синдромы МЭН классифицируют по основным клиническим проявлениям, обусловленным нарушениями секреции гормонов в зависимости от локализации гиперплазии или опухоли эндокринных желез. Нередко встречаются симптомокомплексы, не укладывающиеся в принятую классификацию[6].
- МЭН типа I (синдром Вермера)
- МЭН типа II (синдром Сиппла, МЭН типа IIa)
- МЭН типа III (синдром Горлина, МЭН типа IIb)
МЭН типа I (синдром Вермера)
Включает сочетание гормонально активных опухолей, исходящих из эндокринных клеток и гормонально-неактивных опухолей из неэндокринных клеток организма[2].
Компоненты синдрома Вермера[6]:
- первичный гиперпаратиреоз встречается более чем в 90% случаев, обусловлен гиперплазией нескольких паращитовидных желез. Поражения почек и костей, характерные для гиперпаратиреоза, встречаются не всегда.
- опухоли аденогипофиза отмечаются более чем в 50% случаев. Обычно эти опухоли не гормональноактивны (не секретируют гормоны), наоборот — активно растущие злокачественные опухоли сдавливают и разрушают гипофиз, приводя к пангипопитуитаризму. Реже встречаются аденомы, секретирующие пролактин (персистирующая галакторея и аменорея), СТГ (клиника акромегалии) или АКТГ (гипофизарный синдром Кушинга). СТГ-секретирующая аденома может быть вызвана эктопической продукцией соматолиберина опухолями островковых клеток, надпочечников или злокачественными опухолями из других клеток APUD системы. Удаление эктопических источников соматолиберина позволяет добиться полной регрессии СТГ-секретирующей аденомы.
- опухоли из островковых клеток выявляются более чем в 50% случаев. Обычно они множественные и происходят из разных типов клеток островков (глюкагонома, инсулинома, гастринома, ВИПома).
- опухоли надпочечников — в 40% случаев отмечается гиперплазия или аденома надпочечников. Гиперсекреции глюкокортикоидов обычно не происходит — гиперплазию и аденому находят случайно или при детальном обследовании (КТ).
- заболевания щитовидной железы наблюдаются приблизительно в 20% случаев. Обычно это аденомы или злокачественные опухоли (кроме медуллярной карциномы). Реже встречаются тиреотоксикоз, коллоидный зоб и хронический лимфоцитарный тиреоидит.
- редкие сопутствующие нарушения: рак лёгкого, липомы, полипоз желудка, опухоли яичек, шванномы.
Схема обследования и диагностики[6]:
- определение уровня кальция в сыворотке крови;
- определение уровня пролактина в сыворотке крови;
- определение уровня гастрина и панкреатического полипептида в сыворотке крови;
- выявление гипогликемии (определение уровня глюкозы в крови натощак);
- КТ или МРТ головы для выявления бессимптомных поражений гипофиза.
- гормонально-метаболические исследования: измерение базального уровня инсулина, глюкагона, панкреатического полипептида, соматостатина и гастрина, а также оценка секреции этих гормонов после приёма пищи.
МЭН типа IIa (синдром Сиппла)
Данный синдром обычно включает в себя медуллярную карциному щитовидной железы, феохромоцитому и гиперплазию или аденому паращитовидных желез. Варианты МЭН типа IIa[6]:
- классический вариант МЭН IIa — сочетание медуллярной карциномы щитовидной железы, феохромоцитомы и гиперплазии или аденомы паращитовидных желез;
- семейная изолированная медуллярная карцинома щитовидной железы — данный вариант редко включает другие опухоли;
- МЭН типа IIa с первичным амилоидозом кожи — красно-коричневые, мучительно зудящие высыпания (пятна, узелки) между лопатками или на голенях;
- МЭН типа IIa с болезнью Гиршпрунга.
Диагностическая схема обследования[6] (ближайших родственников пациента или при подозрении):
- определение базального уровня кальция и тирокальцитонина в плазме крови;
- стимуляционные пробы с пентогастрином или кальцием — позволяют выявить гиперплазию C-клеток и медуллярную карциному щитовидной железы;
- определение уровня катехоламинов, ванилил-миндальной кислоты и метанефринов (выявление феохромоцитомы);
- определение уровня АКТГ;
- выявление раково-эмбрионального антигена позволяет диагностировать медуллярную карциному щитовидной железы, если уровень тирокальцитонина не повышен.
МЭН типа IIb (синдром Горлина, МЭН — III)
В данный синдром вошли те же опухоли, что и в МЭН IIa, однако первичный гиперпаратиреоз проявляется реже, а медуллярная карцинома щитовидной железы протекает крайне злокачественно. Выявляется в возрасте 4…37 лет. Отличается от МЭН IIa наличием нейрином (невром) слизистых оболочек и расстройств опорно-двигательного аппарата (марфаноподобная внешность, искривление позвоночного столба, грудной клетки, наличие симптома конской стопы, вывихов головок бедренных костей, арахнодактилия). Нейриномы чаще локализуются на губах, щеках, языке, других отделах желудочно-кишечного тракта и представляют собой бело-розовые безболезненные узелки диаметром от 1…3 мм до 1 см[2].
Клиническая картина
Внешний вид пациентов — характерен прогнатизм, значительное утолщение губ («негроидный» вид), нейриномы слизистых оболочек, нарушения скелета, арахнодактилия.
Клиническая картина определяется наличием соответствующих опухолей[6]:
- медуллярная карцинома щитовидной железы — присутствует практически у всех пациентов. Чем раньше проведена тиреоидэктомия, тем лучше прогноз;
- феохромоцитома встречается приблизительно в 30% случаев;
- первичный гиперпаратиреоз встречается менее 5% случаев;
- множественные нейриномы (невромы) слизистых оболочек обнаруживают более чем в 95% случаев. Нейриномы особенно заметны на губах, веках и языке, придавая лицу характерный вид;
- Нейриномы
 Нейрофиброма подслизистой оболочки губы 13-летнего пациента (синдром МЭН типа IIb)
Нейрофиброма подслизистой оболочки губы 13-летнего пациента (синдром МЭН типа IIb)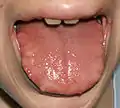 Нейрофибромы края языка 23-летней пациентки с синдромом МЭН типа IIb
Нейрофибромы края языка 23-летней пациентки с синдромом МЭН типа IIb Видны нервные волокна роговицы у 27-летней пациентки с синдромом МЭН типа IIb
Видны нервные волокна роговицы у 27-летней пациентки с синдромом МЭН типа IIb
- другие частые проявления МЭН типа IIb: марфаноподобная внешность, прогерия, воронкообразная деформация грудной клетки, утолщение нервов роговицы, ганглионевромы желудочно-кишечного тракта, мегаколон. Утолщение нервов роговицы иногда наблюдают и при МЭН IIa.
- симптоматика со стороны желудочно-кишечного тракта (диарея, нарушения перистальтики, запоры) может возникать в раннем детском возрасте и быть первым признаком МЭН типа IIb.
Диагностика
- УЗИ щитовидной железы;
- определение уровня тирокальцитонина (пентагастрировый тест), кальция и CEA в крови, экскрецию ванилил-миндальной и гомованилиновой кислоты, адреналина, норадреналина, паратгормона;
- генетический скрининг (мутации RET-онкогена) — обязательное обследование родственников первой и второй степени родства.
Лечение
- Начинают с удаления феохромоцитомы.
- Затем проводят экстирпацию щитовидной железы с удалением шейных лимфатических узлов и клетчатки.
- Назначают препараты тироксина для компенсации послеоперационного гипотиреоза.
- После этого приступают к лечению гиперпаратиреоза и других отклонений.
В неоперабельных случаях и если операция выполнена не радикально — назначают октреотид (синтетический аналог соматостатина) и химиотерапевтические препараты.[6]
Прогноз
При синдроме множественной эндокринной неоплазии определяется степенью злокачественности медуллярной карциномы щитовидной железы, благоприятный прогноз маловероятен[2].
Примечания
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 238-241. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- Carney J.A. Familial multiple endocrine neoplasia: the first 100 years. // Amer. J. Surg. Pathol.— 2005,— Vol. 29, № 2, P.— 254—274.
- Donis-Keller H., Dou S., Chi D., Carlson K.M., Toshima K., Lairmore T.C., Howe J.R., Moley J.F., Goodfellow P., Wells S.A. Jr. (1993) Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. // Hum Mol Genet 2(7), P.— 851—856.
- Guru S.C., Manickam P., Crabtree J.S., Olufemi S.E., Agarwal S.K., Debelenko L.V. Identification and characterization of the multiple endocrine neoplasia type 1 (MEN1) gene. // J. Intern Med.— Vol. 243, № 6, P.— 433—439.
- Эндокринология / Под ред. Н. Лавина. — 2-е изд. Пер. с англ. — М.: Практика, 1999. — С. 891—897. — 1128 с. — 10 000 экз. — ISBN 5-89816-018-3.
См. также
Ссылки
| |||||||||||||||||||||||
| |||||||||||||||||||||||
| |||||||||||||||||||||||
| |||||||||||||||||||||||
Синдромы в эндокринологии | |
|---|---|
| Эпифиз |
|
| Гипоталамус |
|
| Гипофиз |
|
| Щитовидная железа |
|
| Надпочечники |
|
| Половые железы |
|
| Паращитовидные железы | |
| Островки Лангерганса | |
| Диффузная нейроэндокринная система |
|
| Прочие |
|