Синдром де Тони — Дебре — Фанкони
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни (перви́чный изоли́рованный синдро́м Фанко́ни, глюко́зо-фосфа́т-ами́новый диабе́т) — врождённое заболевание, наследуется по аутосомно-рецессивному типу[1]. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната[2]. Одно из рахитоподобных заболеваний.
| Синдром де Тони — Дебре — Фанкони | |
|---|---|
| МКБ-11 | GB90.42 |
| МКБ-10 | E72.0 |
| МКБ-9 | 270.0 |
| OMIM | 613388, 615605 и 134600 |
| DiseasesDB | 11687 |
| MedlinePlus | 000333 |
| eMedicine | ped/756 |
| MeSH | D005198 |
История
Данная тубулопатия была идентифицирована швейцарским педиатром Фанкони среди ранее описанных другими исследователями отдельных частей заболевания. В 1931 году он описал у ребёнка с карликовостью и рахитом глюкозурию и альбуминурию, два года спустя де Тони добавил к клинической картине гипофосфатемию, а вскоре Дебре описал аминоацидурию.
Этиология
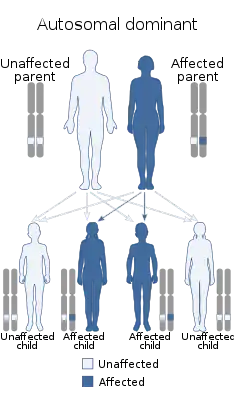

Чаще всего синдром является компонентом других наследственных болезней: цистиноз, тирозинемия типа I, галактоземия, болезнь Вильсона, непереносимость фруктозы. Семейные варианты синдрома наследуются аутосомно-рецессивно, аутосомно-доминантно либо сцепленно с X-хромосомой[2].
Тип наследования — аутосомно-рециссивный, выделена также аутосомно-доминантная форма с локализацией гена на хромосоме 15q15.3. Экспрессивность мутантного гена в гомозиготном состоянии значительно варьирует. Встречаются спорадические случаи, обусловленные свежей мутацией. Считается, что в основе болезни лежат генетически обусловленные дефекты ферментативного фосфорилирования в почечных канальцах (комбинированная тубулопатия), дефицит ферментов 2-го и 3-го комплексов дыхательной цепи — сукцинатдегидрогеназного и цитохромоксидазного. Учёные относят заболевание к разряду митохондриальных болезней.
Патогенез
Патологические изменения представляют собой один из вариантов вторичного гиперпаратиреоза. Основное звено патогенеза — митохондриальный ферментный дефект в цикле Кребса, ферментная тубулопатия, характеризующаяся нарушением реабсорбции глюкозы, аминокислот, фосфатов и бикарбонатов в канальцах почек[1]. Потеря аминокислот и бикарбоната способствует развитию метаболического ацидоза, на фоне которого усиливается резорбция костной ткани и снижается реабсорбция калия и кальция в канальцах почек, что приводит к развитию гипокалиемии и гиперкальциурии. Потеря фосфора ведёт к развитию рахита, а у детей старшего возраста и взрослых — к остеомаляции[2].
Таким образом, митохондриальный ферментный дефект в цикле Кребса ведёт к нарушению процессов энергообеспечения реабсорбции фосфатов, глюкозы и аминокислот в почечных канальцах и повышенной их экскреции с мочой — нарушается кислотно-основное равновесие, а метаболический ацидоз и недостаток фосфатов способствуют разрушению костной ткани по типу рахитоподобных изменений скелета и остеомаляции.
Клиническая картина
Первые признаки заболевания появляются во втором полугодии жизни — дети вялые, гипотрофичные, аппетит резко снижен, наблюдаются рвота, субфебрилитет, гипотония, жажда, полиурия, дегидратация[1]. Развёрнутый симптомокомплекс формируется ко второму году жизни. Если заболевание манифестирует в 5—6 лет, то первыми признаками являются симптомы остеомаляции, деформация костей и гипокалиемические параличи[2]. Со второго года жизни выявляют отставание физического и интеллектуального развития, происходит генерализованная декальцификация[1], проявляющаяся костными деформациями ног (вальгусные или варусные), грудной клетки, предплечий и плечевых костей, снижение мышечного тонуса. Рентгенологически выявляют деформации костей, позвоночного столба, переломы[1], системный остеопороз различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от паспортного возраста ребёнка. Кости становятся ломкими.
При лабораторном обследовании выявляют нормо- или гипокальциемию, гипофосфатемию, повышенный уровень щелочной фосфатазы. В результате снижения реабсорбции бикарбонатов в канальцах почек наблюдается гиперхлоремический ацидоз на фоне избытка паратгормона и нормо- или гипокальциемии. В биохимическом анализе мочи обнаруживают аминоацидурию, глюкозурию (при нормальных уровнях гликемии), натрийурию, гипокальцийурию на фоне гиперфосфатурии[1].
В зависимости от тяжести клинических проявлений и метаболических расстройств выделяют два клинико-биохимических варианта болезни:
- Первый характеризуется значительной задержкой физического развития, тяжёлым течением заболевания с выраженными костными деформациями и нередко переломами костей, резкой гипокальциемией (1,6—1,8 ммоль/л), снижением абсорбции кальция в кишечнике.
- При втором варианте отмечают умеренную задержку физического развития, лёгкое течение с незначительными костными деформациями, нормокальциемию и нормальное усвоение кальция в кишечнике.
Биохимические нарушения
- снижение уровня кальция в крови;
- снижение уровня фосфора в крови;
- повышение уровня щелочной фосфатазы;
- развитие метаболического ацидоза (рН: 7,35…7,25; ВЕ: −10…-12 ммоль/л) за счёт дефекта реабсорбции бикарбонатов в проксимальных канальцах;
- нормальная экскреция кальция с мочой;
- повышение клиренса фосфатов мочи, всасывание фосфатов в кишечнике не страдает;
- развитие глюкозурии (20-30 г/л и выше);
- развитие генерализованной гипераминоацидурии;
- нарушение функций аммониоацидогенеза — снижение титрационной кислотности, повышение рН мочи больше 6,0;
- развитие гипокалиемии.
Исходом заболевания является развитие хронической почечной недостаточности[1].
Дифференциальный диагноз
Дифференциальную диагностику синдрома де Тони-Дебре-Фанкони проводят с рахитом и рахитоподобными заболеваниями у детей[1]. Также дифференцируют со вторичным синдромом, развивающимся на фоне других наследственных и приобретённых заболеваниях:
- синдроме Лоу,
- ювенильном нефронофтизе,
- цистинозе,
- тирозинемии,
- галактоземии,
- гликогенозах,
- наследственной непереносимости фруктозы,
- Rod-cone дистрофии,
- гепатобилиарной дистрофии,
- миеломной болезни,
- амилоидозе,
- синдроме Шегрена,
- нефротическом синдроме,
- почечной трансплантации,
- гиперпаратиреозе, поражении почек солями тяжёлых металлов,
- отравлении лекарственными веществами, в том числе витамином D, лизолом и т. д.).
Лечение
Основные принципы — коррекция электролитных нарушений, сдвигов в кислотно-щелочном равновесии, устранение дефицита калия и бикарбонатов. Увеличивают потребление фосфора с пищей, ограничивают потребление продуктов, включающих серосодержащие аминокислоты, назначают большие дозы витамина D. Для лечения цистиноза с целью подавления накопления цистина в тканях и проксимальных почечных канальцах применяют меркаптамин[2]. Назначают препараты кальция и витамина D, при хронической почечной недостаточности проводится гемодиализ[1].
См. также
- Тубулопатии
- Синдром Барттера
- Fanconi Syndrome
- Zespół Fanconiego
Примечания
- Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 340. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 165—166. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
Литература
- Е. В. Туш — Рахит и рахитоподобные заболевания, Н.Новгород, 2007, с.63-66