Синдром Прадера — Вилли
Синдром Пра́дера — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование. Частота встречаемости — 1 : 12 000-15 000 живорождённых младенцев. Патология встречается с одинаковой частотой у женщин и мужчин[2].
| Синдром Прадера — Вилли | |
|---|---|
 | |
| МКБ-11 | LD90.3 |
| МКБ-10 | Q87.1 |
| МКБ-10-КМ | Q87.1 |
| МКБ-9 | 759.81 |
| МКБ-9-КМ | 759.81[1] |
| OMIM | 176270 |
| DiseasesDB | 10481 |
| MedlinePlus | 001605 |
| eMedicine | ped/1880 |
| MeSH | D011218 |
Наиболее частой причиной синдрома (70-75 % случаев) является делеция участка 15q11-13 хромосомы 15, унаследованной от отца. Около четверти случаев обусловлено однородительской дисомией хромосомы 15 upd(15)mat, когда обе 15-е хромосомы у пациента являются копиями материнского происхождения. В незначительном числе случаев синдром связан с нарушением импринтинга или с наличием сбалансированной транслокации с точкой разрыва внутри участка 15q11-13[3].
Синдром впервые описали в 1956 году учёные из Швейцарии А. Прадер (A. Prader), Х. Вилли (H. Willi) и А. Лабхарт (A. Labhart)[4].
Клиническая картина
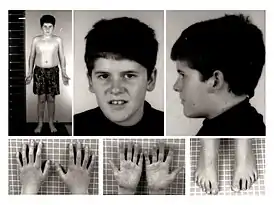
Для синдрома Прадера — Вилли характерны:
- до рождения — низкая подвижность плода, часто — неправильное положение плода;
- дисплазия тазобедренных суставов;
- ожирение; склонность к перееданию (чаще проявляется к двум годам);
- пониженный мышечный тонус (гипотонус); пониженная координация движений;
- маленькие кисти и стопы, низкий рост;
- повышенная сонливость;
- страбизм (косоглазие);
- сколиоз (искривление позвоночника);
- пониженная плотность костей;
- густая слюна; плохие зубы;
- сниженная функция половых желёз (гипогонадизм) и в результате, как правило, бесплодие;
- речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики.
- более позднее половое созревание.
Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы тонкие.
Как правило, у больного встречается не более пяти вышеуказанных признаков.
Диагностика
Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом). Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще).
Дети с синдромом Прадера — Вилли очень похожи между собой, опытный генетик сможет быть уверен в диагнозе, не дожидаясь результатов исследования кариотипа.
Лечение
Синдром Прадера — Вилли является врождённой генетической аномалией; в настоящее время специфические способы его лечения не разработаны. Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии. Рекомендуются использование специальных методик развития ребёнка, занятия с логопедом и дефектологом. Показан приём «гормонов роста», заместительная гормональная терапия (с применением гонадотропинов).
Гипогонадизм обычно проявляется в микропенисе (микропенис — половой член аномально малого размера) и неопущении яичек у мальчиков (крипторхизм); врачи могут посоветовать подождать, пока яички опустятся сами, или порекомендовать хирургическое вмешательство либо гормонотерапию.
Для коррекции избыточной массы тела применяется диета с ограничением количества жиров и углеводов. Из-за ожирения, сопутствующего синдрому, нужно пристально следить за количеством и качеством пищи, поглощаемой человеком с синдромом Прадера — Вилли (обычно люди с таким синдромом способны много съесть, не наедаясь).
Возможным осложнением может стать апноэ (задержка дыхания во сне).
Риск
Риск, что следующий ребёнок у тех же родителей родится также с синдромом Прадера — Вилли, зависит от механизма, вызвавшего генетический сбой.
Этот риск меньше 1 %, если у первого ребёнка делеция гена или партеногенетическая (однородительская) дисомия; составляет до 50 %, если сбой вызван мутацией; до 25 % — в случае транслокации родительских хромосом. Родителям рекомендуется пройти генетическое обследование.
Перспективы развития
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития. Согласно исследованиям, которые провели Л. М. Керфс и Дж. П. Фринс (1992)[5],
- 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ);
- 27 % — уровень на грани среднего (70-85 баллов);
- 34 % — уровень слабого отставания (50-70 баллов);
- 27 % — уровень среднего отставания (35-50 баллов);
- 5 % — сильное отставание (20-35 баллов);
- менее 1 % — значительное отставание.
По другим исследованиям (Кэссиди), 40 % пациентов с синдромом Прадера — Вилли демонстрируют интеллект на грани среднего или сниженный уровень интеллекта.
Как правило, дети с синдромом Прадера — Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарём, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.
Синдром Прадера — Вилли нередко ассоциируется с повышенным аппетитом. У больных повышена концентрация в крови гормона грелина. Для них также характерна пониженная концентрация соматолиберина. Это обусловлено тем, что 15-я хромосома связана с гипоталамусом. Однако при вскрытии умерших с синдромом Прадера — Вилли не было обнаружено никаких дефектов гипоталамуса. По другим данным, наблюдалось снижение общего количества клеток и окситоцин-содержащих клеток паравентрикулярных ядер гипоталамуса[6]
См. также
Примечания
- база данных Disease ontology (англ.) — 2016.
- How many people are affected/at risk for Prader-Willi syndrome (PWS)?. web.archive.org (27 августа 2016). Дата обращения: 17 сентября 2019.
- Buiting K. et al. Clinical utility gene card for: Prader-Willi Syndrome (англ.) // European Journal of Human Genetics. — 2014. — P. e1–e3. — doi:10.1038/ejhg.2014.66.
- Prader A., Labhart A., Willi H. Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myatonieartigem Zustand im Neugeborenenalter (нем.) // Schweiz. Med. Wschr. — 1956. — Bd. 1986. — S. 1260—1261.
- L. M. Curfs, J. P. Fryns. Prader-Willi syndrome: a review with special attention to the cognitive and behavioral profile // Birth Defects Original Article Series. — 1992. — Т. 28, вып. 1. — С. 99–104. — ISSN 0547-6844.
- Swaab, D.F. et al. (1995) Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J. Clin. Endocrinol. Metab. 80, 573—579
Ссылки
- Л. З. Казанцева, П. В. Новиков, А. Н. Семячкина, Е. А. Николаева, М. Б. Курбатов, Э. В. Добрынина. Синдром Прадера — Вилли у детей: новое в этиологии, патогенезе и лечении. Московский НИИ педиатрии и детской хирургии Минздрава РФ / Рос. вестник перинатологии и педиатрии. — 1999. — № 4. — С.40-44.
- Сайт «Биология человека»
- Однородительская дисомия на сайте «Биология человека»
- О синдроме на сайте «Эндокринология» (EURODOCTOR.ru — Элитное лечение в Европе)
- Форум родителей детей с этим диагнозом
- Мэтт Ридли. Геном: автобиография вида: В 23 главах. Глава из книги (Matt Ridley. Genome: The Autobiography of a Species in 23 Chapters)
- Подборка статей по теме, общение родителей (форум)
- Польза и вред от гормонов роста (реферат)
- Кармен Сапиенца. Геномный импринтинг // Scientific American. Издание на русском языке. — 1990. — № 12. — С.14-20.