Синдром Ди Георга
Синдром Ди Георга (синдром Ди Джорджа, синдром Ди Джорджи, синдром дисэмбриогенеза 3-4 жаберной дуги, врождённая аплазия тимуса и паращитовидных желёз, синдром 22q11.2, CATCH 22 phenotype[2]) — разновидность идиопатического изолированного гипопаратиреоза; редкое врождённое заболевание. Генетической причиной синдрома Ди Георга является делеция центрального участка длинного плеча хромосомы 22 (22q11.2) размером 1.5-3 млн.п.н. Однако известны случаи, когда при тех же клинических проявлениях имеет место делеция других хромосом — 10р13, 17р13, 18q21 и других. В большинстве случаев делеция происходит во время мейоза при спермато- или овогенезе. Только в 5-10 % случаев дефектная хромосома наследуется по аутосомно-доминантному типу[3]. Характеризуется агенезией или дисгенезом паращитовидных (околощитовидных) желёз, аплазией тимуса (вилочковой железы), приводящей к резкому снижению популяции Т-лимфоцитов и иммунологической недостаточности, врождёнными аномалиями крупных сосудов (дефекты аорты, тетрада Фалло)[4].
| Синдром Ди Георга | |
|---|---|
 Компьютерная томограмма головного мозга пациента с синдромом Ди Георга демонстрирует кальцификацию базальных ганглиев и перивентрикулярное обызвествление. (По материалам Tonelli et al., 2007). | |
| МКБ-11 | LD44.N0 |
| МКБ-10 | D82.1 |
| МКБ-10-КМ | D82.1 |
| МКБ-9 | 279.11, 758.32 |
| МКБ-9-КМ | 279.11[1] |
| OMIM | 188400 |
| DiseasesDB | 3631 |
| eMedicine | med/567 ped/589 derm/716 |
| MeSH | D004062 |
Этиология и патогенез
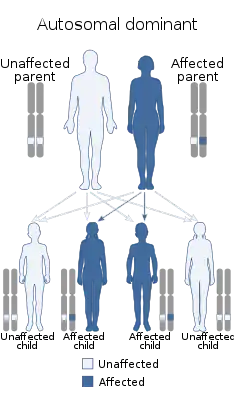
Наиболее вероятная причина развития клинической симптоматики при данном синдроме — несбалансированная транслокация, делеция или микроделеция 22-й хромосомы (22q11.2). Большинство случаев спорадические, обусловлены делециями 22q11[5].
Заболевание развивается в результате повреждения закладки 3-4 жаберных карманов, в результате которого нарушается закладка паращитовидных желёз и тимуса. Тип наследования до конца не установлен — некоторые авторы предполагают аутосомно-доминантный тип с различной экспрессивностью[6].
Клиническая картина
Клинически наиболее постоянными проявлениями заболевания является гипопаратиреоз и кандидомикоз, отмечаются аномалии развития носа, рта, ушей[4].
Заболевание характеризуется аплазией тимуса и связано с нарушениями развития тимуса в эмбриональном периоде. Тимусный эпителий не может обеспечить нормальное развитие Т-клеток. В результате у пациентов с данной формой иммунодефицита страдает как клеточный, так и гуморальный иммунный ответ. Дети с подобным иммунодефицитным заболеванием проявляют повышенную чувствительность к вирусным, грибковым и некоторым бактериальным инфекциям.
Возможно течение синдрома в виде изолированной недостаточности паращитовидных желёз или врождённого отсутствия околощитовидных (паращитовидных) желез — гипокальциемические судороги, начиная с периода новорожденности (тетания) и вилочковой железы (различные инфекционные заболевания как следствие иммунологической недостаточности)[6].
Диагностика
Основывается на выявлении типичных для синдрома аномалий развития[6]:
- агенезия или дисгенез паращитовидных желёз;
- аплазия вилочковой железы;
- иммунологическая недостаточность;
- черепно-лицевые дисморфии (микрогнатия, гипертелоризм, антимонголоидный разрез глаз, расщелины губы и нёба, деформированные и/или низко расположенные ушные раковины).
Наиболее яркие проявления — гипопаратиреоз и кандидомикоз. Возможно сочетание с дефектами аорты и тетрадой Фалло. Иногда — катаракта и паховые грыжи. В крови определяется лимфоцитопения, гипокальциемия, гипогамма-глобулинемия[6].
- Технология распознавания лиц
В Национальном НИИ генома человека в Вашингтоне (США) для диагностики синдрома Ди Джорджи применяется технология распознавания лиц[7].
Прогноз
Обычно больные умирают в раннем возрасте от инфекционных заболеваний и сердечной недостаточности[4].
См. также
- DGCR2 — ассоциированный ген
- Гипокальциемический криз
- Тетания
Примечания
- база данных Disease ontology (англ.) — 2016.
- Burn J. Closing time for CATCH22 (англ.) // J. Med. Genet. : journal. — 1999. — October (vol. 36, no. 10). — P. 737—738. — doi:10.1136/jmg.36.10.737. — PMID 10528851.
- Синдром Ди Джорджи. Центр Молекулярной Генетики при Медико-генетическом научном центре. Дата обращения: 18 марта 2016.
- Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 312. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- Эндокринология / Под ред. Н. Лавина. — 2-е изд. Пер. с англ. — М.: Практика, 1999. — С. 485, 483, 441—442, 66, 64.. — 1128 с. — 10 000 экз. — ISBN 5-89816-018-3.
- Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 67-68. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
- Смит, Браун, 2021, Глава 12, с. 219.
Литература
- Брэд Смит, Кэрол Браун. IT как оружие. Какие опасности таит в себе развитие высоких технологий = Brad Smith, Caroll Ann Browne. Tools and Weapons: The Promise and the Peril of the Digital Age. — М.: Альпина Паблишер, 2021. — 352 с. — ISBN 978-5-9614-4017-1..
Ссылки
| В библиографических каталогах |
|---|
 Медиафайлы по теме Синдром Ди Георга на Викискладе
Медиафайлы по теме Синдром Ди Георга на Викискладе
