GM1-ганглиозидоз
GM1-ганглиозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления. Развитие клинической картины обусловлено дефектом или недостатком β-галактозидазы, который ведёт к нарушению метаболизма и накоплению субстратов (ганглиозида GM1, гликопротеинов и кератансульфата) главным образом в нервных клетках центральной и периферической нервной системы.
| GM1 ганглиозидоз | |
|---|---|
| МКБ-10 | E75.1 |
| МКБ-10-КМ | E75.19 и E75.1 |
| МКБ-9 | 330.1 |
| МКБ-9-КМ | 277.6[1] |
| OMIM | 230600 |
| DiseasesDB | 32008 |
| eMedicine | ped/2891 |
| MeSH | D016537 |
Патогенез
Заболевание характеризуется недостаточностью β-галактозидазы — фермента лизосом, участвующих в катаболизме производных жирных кислот и гликозаминогликанов — ганглиозида GM1, гликопротеинов и кератансульфата.
Бета-галактозидаза является жизненно важным гидролитическим ферментом, обнаруженным в лизосомах, который расщепляет липиды и гликопротеины. В случае генетически обусловленного дефицита или дефекта, когда β-галактозидаза не функционирует должным образом, накапливающиеся в нервной ткани липиды и кератансульфат вызывают проявление характерных клинических симптомов. Большинство вариантов GM1 ганглиозидоза развивается в начале жизни (когда бурно развивается мозг) и сопровождается нейродегенерацией. За исключением редких форм с поздним началом, GM1 ганглиозидозы являются фатальными.
Наследование

Данная группа заболеваний наследуется, как и подавляющее большинство лизосомных болезней накопления, по аутосомно-рецессивному типу наследования[2][3]. Таким образом, с одинаковой частотой встречается как у мужчин, так и у женщин.
Аутосомно-рецессивный тип наследования на практике означает, что дефектный ген расположен на одной из двух гомологичных аутосом. Заболевание клинически манифестирует только в случае, когда обе аутосомы, полученные по одной от отца и матери, являются дефектными по данному гену. Как и во всех случаях аутосомно-рецессивного наследования, если оба родителя несут дефектный ген, то вероятность наследования болезни у потомства составляет 1 из 4. На схеме синим цветом обозначены здоровые, фиолетовым — носители дефектного гена, красным — GM1 ганглиозидоз (два дефектных аллеля GLB1 одного гена 3q21.3). Синим кружочком помечен нормальный аллель, красным — дефектный.
Классификация
Различают три формы заболевания[2]:
- раннюю детскую (инфантильную),
- позднюю детскую (ювенильную),
- взрослая (зрелая).
Согласно Международной классификации болезней десятого пересмотра (МКБ-10), различают:
- E75 Нарушения обмена сфинголипидов и другие болезни накопления липидов.
- E75.1 Другие ганглиозидозы. Ганглиозидоз: БДУ (без дополнительных уточнений), GM1, GM3, Муколипидоз IV.
Ранняя детская форма

Ранняя детская форма GM1 ганглиозидоза — наиболее тяжелая форма данного подтипа ганглиозидоза, которая манифестирует вскоре после рождения ребёнка. Симптомы раннего детского GM1 ганглиозидоза могут включать проявления нейродегенерации, судороги, увеличение размеров печени (гепатомегалия) и селезёнки (спленомегалия), огрубление черт лица, скелетные нарушения, тугоподвижность суставов, вздутие живота, мышечную слабость, чрезмерную реакцию на звук (вздрагивание) и нарушение походки. Приблизительно у половины пациентов развиваются характерные вишнёво-красные пятна на глазном дне. Такие дети по достижении возраста 1 года могут стать слепыми и глухими и часто умирают в возрасте 3 лет от сердечно-сосудистых осложнений или пневмонии.
Среди клинических проявлений данной формы болезни характерно раннее нарушение психомоторного развития ребёнка: снижение активности и вялость в первые недели жизни, проблемы вскармливания — плохая прибавка массы тела. В возрасте 6 месяцев отмечается наличие нистагма, дети не начинают сидеть, а изначальная гипотония впоследствии сменяется развитием спастичности с наличием пирамидных знаков, развивается вторичная микроцефалия, децеребрационная ригидность в 1 год и смерть в возрасте 1-2 года[2] (в результате пневмонии и дыхательной недостаточности).
В некоторых случаях развивается гиперакузия — чрезмерная реакция младенца на звук, проявляющаяся вздрагиванием. В 50 % случаев в возрасте от 6 до 10 месяцев выявляются характерные вишнёво-красные пятна на глазном дне в области макулы, помутнение роговицы. Встречаются признаки лицевого дизморфизма: лобные утолщения, широкая спинка носа, отёк лица (пухлые веки), периферические отёки, эпикант, длинная верхняя губа, микроретрогнатия, гипертрофия дёсен (избыточная толщина альвеолярных гребней), макроглоссия. Обычно гепатомегалия отмечается с 6 месяцев, а спленомегалия развивается позже. У некоторых пациентов отмечаются признаки сердечной недостаточности и деформации скелета: сгибательные контрактуры наблюдаются с 3 месяцев, признаки преждевременного поднадкостничного костеобразования (могут отмечаться у новорождёных), эпифизы формируются позднее, диффузная деминерализация костной ткани, гипоплазия и заострение тел позвонков от грудного до поясничного отдела — в возрасте 3—6 месяцев формируется фиксированный кифоз на месте перехода грудных позвонков к поясничным. Резко выраженная гипоплазия зубовидного отростка может спровоцировать развитие кривошеи и вызвать компрессию спинного мозга различной степени выраженности. Отмечается характерная форма позвонков («рыбьи позвонки») и другие деформации скелета (как в случаях мукополисахаридозов). Внутриклеточное накопление мукополисахаридов напоминает картину синдрома Гурлер: в 10—80 % периферических лимфоцитов отмечаются вакуолизация, в костном мозге — пенистые гистиоциты. Накопление GM1 ганглиозида в сером веществе мозга в 10 раз превышает обычные показатели, а 20—50-кратное увеличение внутренних органов обусловлено внутриклеточным накоплением содержащих галактозу олигосахаридов и умеренного накопления кератансульфата как при синдроме Моркио типа B: мутации с более высокой остаточной активностью бета-галактозидазы в отношении GM1 субстрата, чем для кератансульфата и других гликозаминогликанов, содержащих олигосахариды, которая клинически проявляется минимальными неврологическими нарушениями на фоне значительных деформаций скелета и напоминает синдром Моркио (мукополисахаридоз IV)[3].
Поздняя детская форма
Поздняя детская форма GM1 ганглиозидоза манифестирует позже ранней (обычно в возрасте от 1 до 3 лет). Характеризуется преимущественно неврологической симптоматикой: атаксией, наличием судорог, развитием деменции и расстройствами речи.
Взрослая форма
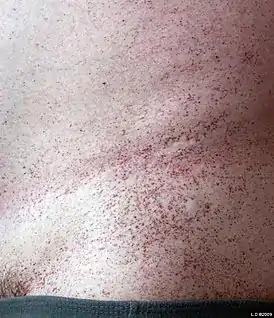
Взрослая форма GM1 ганглиозидоза развивается в возрасте между тремя и тридцатью годами жизни. Клиническая симптоматика характеризуется атрофией мышц, развитием неврологических осложнений, которые, в отличие от детских форм, являются менее тяжёлыми и медленнее прогрессируют, помутнением роговицы (у некоторых пациентов), дистонией (навязчивые мышечные сокращения, которые вызывают торсионную дистонию, повторяющиеся движения или аномальные позы). На нижней части туловища в результате нарушения обмена гликолипидов может развиться ангиокератома. У большинства пациентов отмечаются нормальные размеры печени и селезёнки.
См. также
Примечания
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., Медицина, 1996, 320 с.: ил. Глава 316. Лизосомные болезни накопления (с. 250—273). med-books.info. Дата обращения: 6 января 2015.
- Lyon GL et al., Neurology of Hereditary Metabolic Diseases of Children, ed 2, 1996, p53-55 (англ.)