Болезнь Помпе
Боле́знь Помпе́ (генерализо́ванный гликогено́з, гликогеноз II типа) — редкое наследственное заболевание с аутосомно-рецессивным механизмом наследования[1], связанное с повреждением мышечных и нервных клеток по всему организму. Клиническая картина данной патологии обусловлена накоплением гликогена в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений гликогеноза II типа, в основе всех форм болезни лежит дефицит одного фермента, кодируемого геном GAA.
| Болезнь Помпе | |
|---|---|
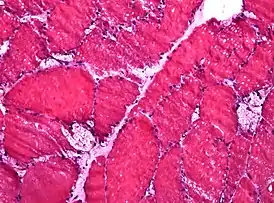 Биопсия мышечной ткани: на замороженном микропрепарате видны крупные вакуоли, характерные для болезни Помпе | |
| МКБ-10 | E74.0 |
| МКБ-10-КМ | E74.02 и E74.0 |
| МКБ-9 | 271.0 |
| OMIM | 232300 |
| DiseasesDB | 5296 |
| eMedicine | med/908 ped/1866 |
| MeSH | D006009 |
Историческая справка
Данное заболевание было впервые описано в 1932 году голландским патологом И. К. Помпе[2][3]. В 1963 году бельгийский биохимик Генри Хэрс обнаружил у пациентов с этой болезнью недостаточность лизосомного фермента α-глюкозидазы[4]. Болезнь Помпе является первым наследственным заболеванием, идентифицированым как лизосомная болезнь накопления, которых сейчас описано около 50[5].
Эпидемиология
Распространённость болезни Помпе (гликогеноза II-го типа, англ. GSD II) составляет примерно 1 случай на 140 000 (классическая инфантильная форма) и 1 на 60 000 взрослого населения (болезнь Помпе с поздним началом)[6]. Заболевание встречается практически во всех этнических группах населения.
Наследование

Заболевание наследуется по аутосомно-рецессивному типу и, таким образом, с одинаковой частотой встречается как у мужчин, так и у женщин.
Ген GAA, мутации в котором вызывают данное заболевание, локализован на аутосоме (хромосоме 17). Заболевание клинически манифестирует только в случае, когда обе аутосомы, полученные по одной от отца и матери, являются дефектными по данному гену. Как и во всех случаях аутосомно-рецессивного наследования, если оба родителя несут дефектный ген, то вероятность наследования болезни у потомства составляет 1 из 4. На схеме синим цветом обозначены здоровые, фиолетовым — носители дефектного гена, красным — болезнь Помпе (два дефектных гена одной аллели 17q25.2-3). Синим кружочком помечен нормальный ген, красным — дефектный.
Патогенез
Генетический дефект локуса 17q25.2-3 хромосомы (ген GAA) ведёт к дефициту кислой α-1,4-глюкозидазы (мальтазы) — одного из ферментов лизосом. В свою очередь дефект этого фермента вызывает прогрессирующее накопление гликогена, которое со временем может стать причиной необратимого поражения мышц и стать причиной смертельного исхода.
Диагностика
Диагноз данной редкой наследственной патологии верифицируется на основании лабораторного исследования степени активности фермента — кислой α-1,4-глюкозидазы (мальтазы) в крови (метод «сухого пятна»)[7] или тканях организма (кожных фибробластах, мышечной ткани, лейкоцитах).
Заболевание в встречается в различных клинических формах:
- классическая (инфантильная) форма болезни Помпе и
- болезнь Помпе с поздним началом (в детском, подростковом возрасте и у взрослых).
Этапы диагностического поиска
Классическая (инфантильная) форма:
- этап: выявление группы риска.
- основные клинические проявления:
- выраженная мышечная гипотония (синдром «вялого ребёнка»);
- задержка моторного развития;
- частые инфекционные заболевания верхних дыхательных путей (ОРЗ);
- наличие признаков дыхательной недостаточности;
- расстройства дыхания во сне;
- кардиомегалия в сочетании с кардиомиопатией;
- сердечная недостаточность;
- макроглоссия;
- гепатомегалия;
- спленомегалия;
- гипотрофия
- лабораторная диагностика: повышение уровня креатинфосфокиназы, лактатдегидрогеназы, АлАТ, АсАТ в сыворотке крови.
- инструментальные методы диагностики:
- поиск наличия признаков поражения миокарда и/или сердечного ритма;
- ЭКГ;
- ЭхоКГ;
- МРТ;
- рентгенография органов грудной полости
- основные клинические проявления:
- этап: специфическая диагностика.
- диагностика: для всех пациентов группы риска болезни Помпе — биохимический анализ активности кислой мальтазы в крови методом сухих пятен
- верификация: повторное измерение уровня активности кислой мальтазы в крови альтернативным методом (например, определение активности этого фермента в лейкоцитах периферической крови)
- этап: дополнительные методы диагностики.
- при наличии типичной клинической картины на фоне «нормальной» активности кислой мальтазы в крови показано проведение ДНК-диагностики дефектного гена (GAA).
- морфологический анализ мышечной ткани (биопсия) позволяет выявить специфические изменения, однако их отсутствие не исключает наличия болезни Помпе.
Диагностический алгоритм болезни Помпе с поздним началом:
- этап: выявление группы риска.
- основные клинические проявления:
- прогрессирующая мышечная слабость тазового и плечевого пояса (на начальных стадиях болезни преобладает слабость в ногах);
- затруднение при ходьбе, подъёме по лестнице, вставании из положения сидя;
- боли в мышцах;
- дыхательная недостаточность;
- ортопноэ;
- частые инфекционные заболевания дыхательных путей;
- дневная сонливость;
- головная боль по утрам;
- ночная гиповентиляция лёгких;
- снижение массы тела у детей и подростков;
- гипотрофия
- лабораторная диагностика: повышение уровня креатинфосфокиназы, лактатдегидрогеназы, АлАТ, АсАТ в сыворотке крови.
- инструментальные методы диагностики:
- выявление миопатического паттерна в ходе проведения миографии скелетных мышц конечностей;
- выявление расстройств дыхания во время сна (эпизоды апноэ и снижение степени оксигенации крови) в ходе проведения полисомнографии;
- выявление значительной разницы результатов измерения форсированной жизненной ёмкости лёгких (ФЖЕЛ) в положении лежа и сидя более, чем на 10 % от исходной величины ФЖЕЛ — ранний признак специфического поражения диафрагмы при болезни Помпе.
- основные клинические проявления:
- этап: специфическая диагностика.
- диагностика: для всех пациентов группы риска болезни Помпе — биохимический анализ активности кислой мальтазы в крови методом сухих пятен
- верификация: повторное измерение уровня активности кислой мальтазы в крови альтернативным методом (например, определение активности этого фермента в лейкоцитах периферической крови)
- этап: дополнительные методы диагностики.
- при наличии типичной клинической картины на фоне «нормальной» активности кислой мальтазы в крови показано проведение ДНК-диагностики дефектного гена (GAA).
- морфологический анализ мышечной ткани (биопсия) позволяет выявить специфические изменения, однако их отсутствие не исключает наличия болезни Помпе.
Клиническая картина
Накопление гликогена ведёт к развитию прогрессирующей мышечной слабости (миопатии). В патологический процесс вовлекаются различные органы и ткани макроорганизма: сердце, скелетные мышцы, печень и нервная система.
Встречаются исключения, однако уровень активности кислой α-1,4-глюкозидазы определяет форму гликогеноза II типа (англ. GSD II). В зависимости от возраста, в котором начинают проявляться клинические симптомы, выделяют две формы заболевания: классическую (или инфантильную) и болезнь Помпе с поздним началом — симптомы появляются позже и прогрессируют медленнее[8].
Классическая (инфантильная) форма
Начало клинических проявлений классической формы болезни, как правило, диагностируется в возрасте 4—8 месяцев. Обращает внимание задержка моторного развития ребёнка грудного возраста. При этом внешне мышцы ребёнка выглядят нормально, однако они вялые и слабые: ребёнок плохо держит голову, практически не переворачивается со спины на живот и обратно. По мере прогрессирования заболевания утолщается сердечная мышца и снижается её сократительная способность. Без лечения смерть обычно наступает в результате сердечной недостаточности и слабости дыхательных мышц[8].
Болезнь Помпе с поздним началом
Развитие клинической картины формы гликогеноза II-го типа с поздним началом происходит позже (в возрасте 1—2 лет), прогрессирует медленнее классической (инфантильной) формы. Одним из первых симптомов является прогрессирующее снижение силы мышц. Оно начинается с ног и распространяется на мышцы туловища и рук, в том числе диафрагму и другие мышцы, участвующие в акте дыхания. Наиболее распространённой причиной смерти является дыхательная недостаточность. В некоторых случаях отмечается увеличение размеров сердечной мышцы и наблюдаются нарушения сердечного ритма, однако это не относится к постоянным признакам данного заболевания[8].
Дифференциальная диагностика
Диагноз устанавливается с задержкой, поскольку признаки и симптомы заболевания весьма похожи на проявления других болезней. Тем не менее, ранняя диагностика и специфическое лечение крайне необходимы для улучшения состояния пациента.
Дифференциальная диагностика гликогеноза II-го типа проводится:
- с другими лизосомными болезнями накопления: синдром Данона;
- с другими гликогенозами;
- с другими заболеваниями: полиомиозит, мышечная дистрофия Беккера — Дюшена, идиопатическая гипертрофическая кардиомиопатия, конечностно-поясная мышечная дистрофия.
Лечение
Существующие на сегодняшний день лечебные стратегии могут улучшить качество жизни пациентов с гликогенозом II-го типа, однако они не способны изменить течение болезни. Раннее начало терапии чрезвычайно важно для достижения наибольшего эффекта.
Единственный вариант специфического лечения заболевания — терапевтическое замещение повреждённого либо отсутствующего фермента фармацевтическим препаратом «Майозайм» (алглюкозидаза альфа[9], rhGAA). Данный препарат разработан компанией «Genzyme Corp.» и успешно прошёл клинические испытания, в которых участвовало 39 больных в возрасте от 1 месяца до 3.5 лет. В 2006 году он был одобрен FDA для лечения пациентов с инфантильной формой болезни в США[10]. Препарат «Майозайм» относится к одним из самых дорогих лекарств в мире, и годовой курс обходится от 100 до 300 тысяч долларов США в зависимости от возраста и веса пациента[11]. C 2013 года препарат зарегистрирован в РФ[12].
В 2021 г. был одобрен препарат авалглюкозидаза альфа.[13]
Прогноз
Прогноз данного редкого генетически детерминированного заболевания варьирует в зависимости от времени наступления и степени выраженности клинических симптомов. Без своевременного специфического лечения эта генетическая патология, особенно среди младенцев и маленьких детей, часто заканчивается летально.
См. также
Примечания
- Pompe disease at NLM Genetics Home Reference (англ.)
- Pompe, J.C. Over idiopathische hypertrophie van het hart (нид.) // Nederlands Tijdschrift voor Geneeskunde. — 1932. — Bd. 76. — P. 304—312. (англ.)
- of Glycogen-Storage Disease Type II (Pompe Disease) 947870, раздел Genetics of Glycogen-Storage Disease Type II (Pompe Disease) (англ.) на сайте EMedicine
- Hers H. G. α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe's disease) (англ.) // Biochemical Journal. — 1963. — Vol. 86, no. 1. — P. 11—16. — PMID 13954110.
- la Marca G. Lysosomals // Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases / N. Blau, M. Duran, K. M. Gibson, C. D. Vici. — Springer Berlin Heidelberg, 2014. — P. 785-793. — ISBN 978-3-642-40336-1. (англ.)
- Ausems M.G., Verbiest J., Hermans M.P., et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling (англ.) // European Journal of Human Genetics : journal. — 1999. — September (vol. 7, no. 6). — P. 713—716. — doi:10.1038/sj.ejhg.5200367. — PMID 10482961. (англ.)
- Umapathysivam K., Hopwood J.J., Meikle P. J. Determination of acid alpha-glucosidase activity in blood spots as a diagnostic test for Pompe disease. Clin. Chem. 2001; 47; 1378—1383. (англ.)
- Type II Glycogen Storage Disease (недоступная ссылка). The Association for Glycogen Storage Disease. Дата обращения: 27 ноября 2014. Архивировано 23 июня 2012 года. (англ.)
- LUMIZYME- alglucosidase alfa injection, powder, for solution (англ.). DailyMed. U. S. National Library of Medicine.
- FDA Approves First Treatment for Pompe Disease (англ.). U.S. Food and Drug Administration (28 апреля 2006). Дата обращения: 15 апреля 2015.
- Matthew Herper. The World's Most Expensive Drugs (англ.). Forbes.com LLC (22 февраля 2010). Дата обращения: 15 апреля 2015.
- Государственный реестр лекарственных средств. grls.rosminzdrav.ru. Дата обращения: 14 января 2016.
- NEXVIAZYME NGPT- avalglucosidase alfa injection, powder, lyophilized, for solution (англ.). DailyMed. U. S. National Library of Medicine.