Наследственная оптическая нейропатия Лебера
Наследственная оптическая нейропатия LHON Лебера, или атрофия зрительного нерва Лебера , является наследственной (передаётся от матери к потомству) митохондриальной дегенерацией ганглионарных клеток (РСК) сетчатки и их аксонов, что приводит к острой или почти острой потере центрального зрения; это влияет преимущественно на молодых мужчин. Тем не менее, LHON передается только по материнской линии, прежде всего, из-за мутаций (не ядерных) в митохондриальном геноме, и только яйцеклетка способствует митохондрии в зародыше. LHON, как правило, связана с одной из трех патогенных митохондриальных ДНК (мтДНК) точечных мутаций. Эти мутации действуют на нуклеотиды и репозиционируют 11778 G в А , 3460 G в А и 14484 T в C, соответственно, в ND4, ND1 и Nd6 субъединицах генов в комплексе I окислительного фосфорилирования цепочек в митохондриях. Мужчины не могут передать болезнь своему потомству.[1]
| Наследственная оптическая нейропатия Лебера | |
|---|---|
| МКБ-10 | H47.2 |
| МКБ-10-КМ | H47.22 и H47.2 |
| МКБ-9 | 377.16 |
| OMIM | 535000 |
| DiseasesDB | 7340 |
| MeSH | D029242 |
История
Это заболевание было впервые описано немецким офтальмологом Теодором Лебером (1840—1917) в 1871 году.[2] В своей статье Лебер описал четыре семьи в которых молодые люди страдали от резкой потери зрения в обоих глазах одновременно или последовательно. Первоначально считалось, что заболевание было связано с Х-хромосомой, но впоследствии было доказано, что оно носит митохондриальный характер[3]. Мутация была впервые идентифицирована в 1988 году Уоллесом и др., которые обнаружили замены нуклеотидов гуанина (G) в аденозина (А) в позиции 11778 в девяти семьях[4]. Эта мутация преобразует высоко консервативный аргинин-340 НАДН-дегидрогеназы, комплекса I в митохондриальной дыхательной цепи, в гистидин. Две другие мутации, вызывающие это заболевание, были идентифицированы в 1991 году (замена G на A в позиции 3460)[5] и 1992 году (замена тимидина (Т) на цитозин (С) в позиции 14484)[6]. Эти три мутации составляют свыше 95 % случаев: мутация по позиции 11778 — 50-70 % случаев, мутации 14484 — 10-15 %, а мутация 3460 — 8-25 %.
Признаки и симптомы
Клинически — острое начало потери зрения, сначала в одном глазу, а затем через промежуток времени от нескольких недель до нескольких месяцев — в другом. Начинается обычно в юности, но были сообщения о начальном возрасте в диапазоне 7-75 лет. Начальный возраст немного выше у женщин, (диапазон 19-55 лет: в среднем 31,3 лет), чем мужчин (диапазон 15-53 лет: в среднем 24,3 года). Соотношение между мужчинами и женщинами колеблется в зависимости от мутаций: 3: 1 для 3460 G>A, 6: 1 для 11 778 G>А и 8: 1 для 14484 T>C.
Это, как правило, развивается в очень тяжелую атрофию зрительного нерва и постоянное снижение остроты зрения, отражаясь на оба глаза одновременно (25 % случаев) или последовательно (75 % случаев) со средним интервалом 8 недель. Лишь в редких случаях только один глаз может пострадать. В острой стадии, длящейся несколько недель, пораженный глаз демонстрирует появление отёка слоя нервных волокон, особенно в дугообразных пучках и увеличенных или телеангиэктатических и извилистых околососковых сосудов (микроангиопатии). Главные особенности видны при офтальмоскопии, перед или после потери зрения. Дефекты зрачка также могут быть видны в острой стадии. Анализ показывает снижение остроты зрения, потерю цветового зрения и тифлоцентральную скотому при тестировании поля зрения.
LHON Плюс
«LHON Плюс» — название, данное редким случаям заболевания при наличии других патологических состояний.[7] Симптомы этой высшей формы заболевания включают потерю способности мозга контролировать движения мышц, тремор, и сердечную аритмию.[8] Во многих случаях LHON Плюс сравнивался с рассеянным склерозом из-за нарушения работы мышц, связанными с нарушениями работы головного мозга.[9]
Генетика
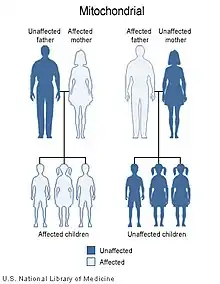
Наследственная оптическая невропатия Лебера является состоянием, связанным с изменениями в митохондриальной ДНК . Хотя большинство ДНК упаковано в хромосомах внутри ядра, митохондрии имеют особый митохондриальный геном, состоящий из мтДНК.
Мутации генов МТ-ND1 , МТ-ND4 , МТ-ND4L и MT-Nd6 вызывают наследственную оптическую невропатию Лебера.[10] Эти гены кодируют мембранную часть белка НАДН-дегидрогеназы, участвующего в нормальной митохондриальной функции окислительного фосфорилирования . Окислительное фосфорилирование использует серию из четырех крупных мультиферментных комплексов, которые все встроены во внутреннюю мембрану митохондрий для преобразования кислорода и моносахаридов в энергию. Мутации в любом из генов нарушают этот процесс, вызывая различные синдромы в зависимости от типа мутации и других факторов. Остается неясным, как эти генетические изменения приводят к гибели клеток в зрительном нерве и к другим специфическим симптомам наследственной оптической нейропатии Лебера.
Эпидемиология
У населения Северной Европы примерно у одного из 9000 человек имеется один из трех основных видов мутаций LHON.[11][12] Распространённость заболевания в Европе составляет от 1: 30000 до 1: 50000.
Мутация LHON ND4 G11778A доминирует в качестве основной мутации в большинстве стран мира с 70 % случаев Северной Европы и 90 % случаев азиатских стран. Из-за эффекта основателя, мутация LHON T14484C ND6 происходит в 86 % случаев LHON в Квебеке,Канада .[13]
Более 50 процентов мужчин и более 85 процентов женщин с мутацией никогда не испытывают потерю зрения или связанных с ней медицинских проблем. Конкретный тип мутации может предсказать вероятность пенетрантности, тяжесть заболевания и вероятность восстановления зрения у пострадавших. Как правило, женщина, которая гомоплазмически вынашивает, основная LHON мутация имеет ~ 40 % риск появления больного сына и ~ 10 % риск появления больной дочери.
Дополнительные факторы могут определить, развиваются ли у человека признаки и симптомы этого расстройства. Экологические факторы, такие как курение и употребление алкоголя могут быть использованы, хотя исследования этих факторов, дали противоречивые результаты. Исследователи также изучают изменения дополнительных генов, в частности генов на Х-хромосоме,[14][15] их вклад в развитие признаков и симптомов. Степень гетероплазмии, процент митохондрий, которые имеют мутантные аллели, могут также играть определенную роль.[16] Модели митохондриальных аллелей называемых гаплогруппами также может влиять на экспрессию мутаций.[17]
Патофизиология
Глазная патология ограничивается слоем ганглиозных клеток сетчатки особенно макулопапилярного узла. Вырождение очевидно от ганглиозных клеток органов сетчатки до аксональных путей, ведущих к боковому латеральному коленчатому телу . Экспериментальные данные показывает нарушение транспорта глутамата и увеличение активных форм кислорода (АФК), вызывающие апоптоз ганглиозных клеток сетчатки. Кроме того, опыты показывают, что обычно, без LHON, ганглиозные клетки сетчатки производят меньше сильнодействующего свободнорадикального супероксида, чем другие обычные нейроны центральной нервной системы.[18] В экспериментах вирусного вектора, увеличивающих супероксиддисмутаза 2 в LHON цибридах[19] или LHON моделей животных или использования экзогенного глутатиона в LHON цибридах[20] было показано, что существует риск гибели пострадавших от LHON ганглиозных клеток сетчатки от апоптоза. Эти эксперименты могут частично объяснить предпочтительность смерти пострадавших от LHON ганглиозных клеток сетчатки другим нейронам центральной нервной системы, которые также несут пострадавшие от LHON митохондрии.
Диагностика и лечение
Без знания истории семьи LHON диагноз, как правило, требует нервно-офтальмологической оценки и анализа крови для митохондриальной оценки ДНК.[21] Здесь важно исключить влияние других возможных причин потери зрения и важных, связанных синдромов, таких как сердечная электрическая проводимость системных аномалий. Прогноз для пострадавших, остающихся неизлечимыми, почти всегда означает продолжение существенного снижения зрения в обоих глазах. Регулярные проверки остроты зрения и проверки периметрии рекомендуется для дальнейших шагов пострадавших лиц. Существует терапия для некоторых случаев этого заболевания, особенно для раннего начала болезни.[22] Кроме того, экспериментальные протоколы лечения продолжаются.[23] Должно быть предложено генетическое консультирование . Здоровье и образ жизни должны быть пересмотрены, особенно в свете токсичных и пищевых теорий экспрессии генов. Зрячие помощники и восстановительные работы должны быть использованы для оказания помощи в сохранении рабочих мест.
Для тех, кто являются носителями мутации LHON, доклинические маркеры могут быть использованы для мониторинга прогресса.[24] Например фотография дна может контролировать отёк слоя нервных волокон. Оптическая когерентная томография может быть использована для более детального изучения толщины слоя нервных волокон сетчатки. Тестирование красно-зеленого цветоощущения может обнаружить потери. Контрастная чувствительность может быть уменьшена. Может быть выявлена ненормальная электроретинограмма или зрительный вызванный потенциал . Нейрон-энолаза и маркеры аксонов тяжелой цепи нейрофиламентов крови может предсказать статус преобразования для пострадавших.
Цианокобаламин (форма витамина В12) следует избегать, поскольку это может привести к слепоте у больных болезнью Лебера.
Как правило рекомендуется избегать токсинов зрительного нерва, особенно табак и алкоголь. Некоторые отпускаемые по рецепту лекарства, как известно, несут потенциальный риск, так что ко всем препаратам следует относиться с подозрением и проверить перед использованием степень риска. Этамбутол, в частности, был причастен как импульс к потери зрения у носителей LHON. В самом деле, токсичные и пищевые оптические нейропатии могут иметь пересекающиеся с LHON симптомы, митохондриальные механизмы болезни и управления.[25] Следует отметить, когда пациент в результате LHON или токсической/ пищевой оптической нейропатиии перенес гипертонический криз, усложняющий процесс болезни, нитропруссид (торговое название: Nipride) не должны использоваться в связи с повышенным риском ишемии зрительного нерва, как следствие реакции на этот антигипертензивный препарат.[26]
Идебенон[22][27][28] в небольшом плацебо-контролируемом исследовании, был показан примерно половине пациентов для достижения умеренной пользы. Лучшие результаты были у людей, которые были в начале заболевания.
α-Токотриенол-quinone, метаболит витамина Е, имел в небольших открытых исследованиях некоторый успех в обращении вспять начавшейся потери зрения[23][29].
Есть различные подходы к лечению, которые проходили предварительные испытания или предложения, ни один еще не привел убедительных доказательств полезности и безопасности для лечения или профилактики том числе: бримонидин ,[30] Миноциклин ,[31] куркумин ,[32] глутатион ,[20] фототерапия инфракрасного излучения ,[33] и методы вирусного вектора.[19]
«Экстракорпоральное оплодотворение в третьем лице» является доказательством концепции исследовательских методов для предотвращения митохондриальной болезни в развитии человеческого плода. До сих пор, были произведены жизнеспособные макаки. Но препятствия этического и познавательного характера останавливают использование этого метода на людях.[34]
См. также
Примечания
- Bandelt H.J., Kong Q.P., Parson W., Salas A. More evidence for non-maternal inheritance of mitochondrial DNA? (англ.) // Journal of Medical Genetics : journal. — 2005. — December (vol. 42, no. 12). — P. 957—960. — doi:10.1136/jmg.2005.033589. — PMID 15923271.
- Leber T. Ueber hereditaere und congenital angelegte sehnervenleiden (1871) Graefes Arch Clin Exp Ophthalmol. 17:249-291
- Erickson R.P. Leber's optic atrophy, a possible example of maternal inheritance (англ.) // American Journal of Human Genetics : journal. — 1972. — Vol. 24, no. 3. — P. 348—349.
- Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas LJ 2nd, Nikoskelainen E.K. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy (англ.) // Science : journal. — 1988. — Vol. 242, no. 4884. — P. 1427—1430. — doi:10.1126/science.3201231. — PMID 3201231.
- Huoponen K., Vilkki J., Aula P., Nikoskelainen E.K., Savontaus M.L. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy (англ.) // American Journal of Human Genetics : journal. — 1991. — Vol. 48, no. 6. — P. 1147—1153.
- Johns D.R., Neufeld M.J., Park R.D. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy (англ.) // Biochemical and Biophysical Research Communications : journal. — 1992. — Vol. 187, no. 3. — P. 1551—1557. — doi:10.1016/0006-291x(92)90479-5.
- Nikoskelainen E.K., Marttila R.J., Huoponen K., et al. Leber's "plus": neurological abnormalities in patients with Leber's hereditary optic neuropathy (англ.) // Journal of Neurology, Neurosurgery and Psychiatry : journal. — 1995. — August (vol. 59, no. 2). — P. 160—164. — doi:10.1136/jnnp.59.2.160. — PMID 7629530.
- cardiac arrythmia (недоступная ссылка). Дата обращения: 2 декабря 2014. Архивировано 30 января 2020 года.
- Mayo Clinic: Multiple Sclerosis
- OMIM 535000
- Man P.Y., Griffiths P.G., Brown D.T., Howell N., Turnbull D.M., Chinnery P.F. The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England (англ.) // American Journal of Human Genetics : journal. — 2003. — February (vol. 72, no. 2). — P. 333—339. — doi:10.1086/346066. — PMID 12518276.
- Puomila A., Hamalainen P., Kivioja S., et al. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland (англ.) // European Journal of Human Genetics : journal. — 2007. — October (vol. 15, no. 10). — P. 1079—1089. — doi:10.1038/sj.ejhg.5201828. — PMID 17406640.
- Laberge A.M., Jomphe M., Houde L., et al. A "Fille du Roy" Introduced the T14484C Leber Hereditary Optic Neuropathy Mutation in French Canadians (англ.) // American Journal of Human Genetics : journal. — 2005. — Vol. 77, no. 2. — P. 313—317. — doi:10.1086/432491. — PMID 15954041.
- Hudson G., Carelli V., Horvath R., Zeviani M., Smeets H.J., Chinnery P.F. X-Inactivation patterns in females harboring mtDNA mutations that cause Leber hereditary optic neuropathy (англ.) // Molecular Vision : journal. — 2007. — Vol. 13. — P. 2339—2343. — PMID 18199976.
- Hudson G., Keers S., Yu Wai Man P., et al. Identification of an X-Chromosomal Locus and Haplotype Modulating the Phenotype of a Mitochondrial DNA Disorder (англ.) // American Journal of Human Genetics : journal. — 2005. — December (vol. 77, no. 6). — P. 1086—1091. — doi:10.1086/498176. — PMID 16380918.
- Chinnery P.F., Andrews R.M., Turnbull D.M., Howell N.N. Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation? (англ.) // American Journal of Medical Genetics : journal. — 2001. — January (vol. 98, no. 3). — P. 235—243. — doi:10.1002/1096-8628(20010122)98:3<235::AID-AJMG1086>3.0.CO;2-O. — PMID 11169561.
- Hudson G., Carelli V., Spruijt L., et al. Clinical Expression of Leber Hereditary Optic Neuropathy Is Affected by the Mitochondrial DNA-Haplogroup Background (англ.) // American Journal of Human Genetics : journal. — 2007. — August (vol. 81, no. 2). — P. 228—233. — doi:10.1086/519394. — PMID 17668373.
- Hoegger M.J., Lieven C.J., Levin L.A. Differential production of superoxide by neuronal mitochondria (англ.) // BMC Neurosci : journal. — 2008. — Vol. 9. — P. 4. — doi:10.1186/1471-2202-9-4. — PMID 18182110.
- Qi X., Sun L., Hauswirth W.W., Lewin A.S., Guy J. Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutation (англ.) // JAMA : journal. — 2007. — February (vol. 125, no. 2). — P. 268—272. — doi:10.1001/archopht.125.2.268. — PMID 17296905.
- Ghelli A., Porcelli A.M., Zanna C., Martinuzzi A., Carelli V., Rugolo M. Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids (англ.) // Investigative Ophthalmology & Visual Science : journal. — 2008. — February (vol. 49, no. 2). — P. 671—676. — doi:10.1167/iovs.07-0880. — PMID 18235013.
- GeneTests LHON search (недоступная ссылка)
- Klopstock, T.; Yu-Wai-Man, P., Dimitriadis, K., Rouleau, J., Heck, S., Bailie, M., Atawan, A., Chattopadhyay, S., Schubert, M., Garip, A., Kernt, M., Petraki, D., Rummey, C., Leinonen, M., Metz, G., Griffiths, P. G., Meier, T., Chinnery, P. F. A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy (англ.) // Brain : journal. — Oxford University Press, 2011. — 25 July (vol. 134, no. 9). — P. 2677. — doi:10.1093/brain/awr170.
- Shrader W. D., Amagata A., Barnes A., Enns G. M., Hinman A., Jankowski O., Kheifets V., Komatsuzaki R., Lee E., Mollard P., Murase K., Sadun A. A., Thoolen M., Wesson K., Miller G. α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. (англ.) // Bioorganic & medicinal chemistry letters. — 2011. — Vol. 21, no. 12. — P. 3693—3698. — doi:10.1016/j.bmcl.2011.04.085. — PMID 21600768.
- Sadun A.A., Salomao S.R., Berezovsky A., et al. SUBCLINICAL CARRIERS AND CONVERSIONS IN LEBER HEREDITARY OPTIC NEUROPATHY: A PROSPECTIVE PSYCHOPHYSICAL STUDY (англ.) // Trans Am Ophthalmol Soc : journal. — 2006. — Vol. 104. — P. 51—61. — PMID 17471325.
- Carelli V., Ross-Cisneros F.N., Sadun A.A. Mitochondrial dysfunction as a cause of optic neuropathies (англ.) // Prog Retin Eye Res : journal. — 2004. — January (vol. 23, no. 1). — P. 53—89. — doi:10.1016/j.preteyeres.2003.10.003. — PMID 14766317.
- Katz, Jason; Patel, Chetan. Parkland Manual of Inpatient Medicine (неопр.). — Dallas, TX: FA Davis, 2006. — С. 903.
- Clinical Idebenone trial recruiting at Newcastle University UK http://lhon.ncl.ac.uk
- Mashima Y., Kigasawa K., Wakakura M., Oguchi Y. Do idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy? (англ.) // J Neuroophthalmol : journal. — 2000. — September (vol. 20, no. 3). — P. 166—170. — doi:10.1097/00041327-200020030-00006. — PMID 11001192.
- Архивированная копия (недоступная ссылка). Дата обращения: 5 июня 2011. Архивировано 4 сентября 2011 года. Sadun,A et al. «EPI-743 alters the natural history of progression of Leber hereditary optic neuropathy». AOS meeting. May 2011]
- Newman N.J., Biousse V., David R., et al. Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite (англ.) // American Journal of Ophthalmology : journal. — 2005. — September (vol. 140, no. 3). — P. 407—415. — doi:10.1016/j.ajo.2005.03.058. — PMID 16083844.
- Haroon M.F., Fatima A., Scholer S., et al. Minocycline, a possible neuroprotective agent in Leber's hereditary optic neuropathy (LHON): Studies of cybrid cells bearing 11778 mutation (англ.) // Neurobiology of Disease : journal. — 2007. — Vol. 28, no. 3. — P. 237—250. — doi:10.1016/j.nbd.2007.07.021. — PMID 17822909.
- Clinical Curcurmin trial recruiting at ClinicalTrials.nlm.nih.gov Архивировано 13 февраля 2009 года.
- Wisconsin near infrared trial Архивировано 15 мая 2008 года.
- Craven L., Tuppen H.A., Greggains G.D., Harbottle S.J., Murphy J.L., Cree L.M., Murdoch A.P., Chinnery P.F., Taylor R.W., Lightowlers R.N., Herbert M., Turnbull D.M. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease (англ.) // Nature : journal. — 2010. — May (vol. 465, no. 7294). — P. 82—85. — doi:10.1038/nature08958. — PMID 20393463.
Ссылки
- Yu-Wai-Man P, Chinnery P (2011) Leber Hereditary Optic Neuropathy. GeneReviews
- Kerrison J.B., Newman N.J. Clinical spectrum of Leber's hereditary optic neuropathy (англ.) // Clin. Neurosci. : journal. — 1997. — Vol. 4, no. 5. — P. 295—301. — PMID 9292259.
- Carelli V., Ross-Cisneros F.N., Sadun A.A. Mitochondrial dysfunction as a cause of optic neuropathies (англ.) // Prog Retin Eye Res : journal. — 2004. — January (vol. 23, no. 1). — P. 53—89. — doi:10.1016/j.preteyeres.2003.10.003. — PMID 14766317.
- LHONsociety.org a Charity Registered with the Charity Commission for England & Wales number 1157206 — The LHON Society was established by the Trustees to create a home for those in the British Isles affected by and with an interest in, LHON. Our aspiration is that through the LHON Society you have access to:
- Shared experiences of those living with LHON and their families
- Sources of practical and emotional support
- Up-to-date information on practical innovation and scientific progress into LHON