Синдром Кернса — Сейра
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьезным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии (сокращенно CPEO), синдром, который характеризуется изолированным поражением мышц, контролирующих движения век (поднимающая верхнее веко, круговая мышца глаза) и контролирующих движения глаз (экстраокулярных мышц). Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения[1]. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.
| Синдром Кернса — Сейра | |
|---|---|
| МКБ-10 | H49.8 |
| МКБ-10-КМ | H49.81 и H49.8 |
| МКБ-9 | 277.87 |
| OMIM | 530000 |
| DiseasesDB | 7137 |
| eMedicine | article/950897 |
| MeSH | D007625 |
История
Это триада: CPEO, двусторонняя пигментная ретинопатия и блокада сердца была впервые описана в докладе о случае у двух пациентов в 1958 году доктором медицинских наук Томасом П. Кернс, (англ. Thomas P. Kearns) и доктором медицинских наук Джорджем Помероя Сейр (англ. George Pomeroy Sayre)[2]. Второй случай был опубликован в 1960 году Ягером и соавторами, которые сообщили об этих симптомах у 13-летнего мальчика[3]. Предыдущие случаи внезапной смерти пациентов с CPEO были опубликованы, как от сердечной аритмии. Другие случаи отмечали особую пигментацию сетчатки, но ни одна из этих публикаций не была документирована как три патологии, возникающие вместе в качестве генетического синдрома[4]. Кернс опубликовал определяющий случай в 1965 году, описывающий 9 несвязанных случаев этой триады[4]. В 1988, была впервые выявлена связь между KSS и крупномасштабными удалениями мышечной митохондриальной ДНК (сокращенно мтДНК)[5][6]. После этого открытия, многочисленные делеции в митохондриальной ДНК были связаны с развитием КСС[7][8][9].
Этиология
Синдром Кернса — Сейра происходит спонтанно в большинстве случаев. В некоторых случаях была передача по наследству посредством митохондриального, аутосомно-доминантного, или аутосомно-рецессивного наследования. У него нет пристрастия к расе или полу, и нет никаких известных факторов риска. По состоянию на 1992 было всего 226 случаев, зарегистрированных в опубликованной литературе[10]. По состоянию на 2017г. получены данные о том, что при синдроме Кернса – Сейра не прослеживается наследственная передача данной патологии, заболевание регистрируется в виде единичных случаев.
Признаки и симптомы
Лица с KSS предстают первоначально со сходными симптомами с типичной CPEO. Начало в первой и второй декадах жизни.
Первым симптомом этого заболевания является односторонний птоз, или проблемы при открытии век, который постепенно прогрессирует и приводит к двустороннему птозу. Когда птоз усиливается, пострадавший обычно запрокидывает шею, поднимая подбородок в попытке предотвратить окклюзию зрительной оси опустившимися веками. Наряду с коварным развитием птоза, движения глаз в конечном итоге становятся ограниченными, в результате чего, лицо больше полагается на поворот головы из стороны в сторону или вверх и вниз для просмотра объектов в периферическом поле зрения.
- Пигментная ретинопатия
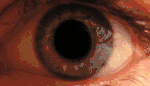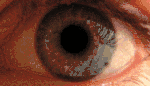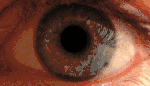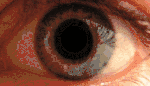
KSS приводит к пигментации сетчатки, прежде всего, в задней части глазного дна. Наблюдается диффузная депигментация пигментного эпителия сетчатки с наибольшим эффектом в жёлтом пятне. Этим KSS отличается от пигментного ретинита, где пигментируется периферия. Вид сетчатки при KSS аналогичен тому, которое наблюдалось при миотонической дистрофии типа 1 (сокращенно DM1). Умеренная куриная слепота может наблюдаться у пациентов с KSS. Потеря остроты зрения, как правило, мягкая и происходит только у 40-50 % больных[11].
- Нарушения сердечной проводимости
Это чаще всего происходит после образования птоза и офтальмоплегии[11]. Атриовентрикулярная блокада (сокращенно «AV») является наиболее распространенным дефицитом сердечной проводимости. Это часто прогрессирует до третьей степени желудочковой блокады, которая является полным блокированием проводимости от предсердия в желудочек. Симптомы сердечной блокады включают обморок, непереносимость физических нагрузок и брадикардию
- Церебральная фолатная недостаточность
У пациентов с синдромом Кернса — Сейра очень часто обнаруживается церебральная фолатная недостаточность — синдром, при котором уровни 5-MTHF в спинномозговой жидкости снижены, несмотря на нормальные уровни фолиевой кислоты и 5-MTHF в плазме крови[12]. Назначение фолиниевой кислоты может в некоторых случаях облегчить симптомы недостаточности и даже скорректировать наблюдаемые на снимках мозга отклонения, особенно если терапия была начата на ранних стадиях заболевания[13]. Предполагаемая причина церебральной фолатной недостаточности у пациентов с синдрмом Кернса-Сейра — дисфункция сосудистого сплетения, нарушающая поступление фолатов в спинномозговую жидкость[14].
- Другие
Согласно описанию заболевания, представленному Кернс в 1965 году, а также описаниям в более поздних публикациях, некоторые симптомы возникают не у всех пациентов. Среди этих симптомов — слабость мышц лица, глотки, туловища и мышц конечностей, потеря слуха, небольшой рост, электроэнцефалографические изменения, мозжечковая атаксия и повышенный уровень белка в спинномозговой жидкости.
Генетика
KSS является результатом делеций в митохондриальной ДНК (мтДНК), которые вызывают определённый фенотип. мтДНК передается исключительно от яйцеклетки матери[15]. Митохондриальная ДНК состоит из 37 генов, найденных в одной кольцевой хромосоме размерностью 16569 спаренных оснований в длину. Среди них 13 генов кодируют белки дыхательной цепи переноса электронов(сокращенно «ЭTЦ»), 22 кодируют транспортировку РНК (тРНК) и два кодируют ряд больших и малых субъединиц, которые образуют рибосомные РНК (рРНК). 13 белков, участвующие в ЭТЦ в митохондриях, необходимы для окислительного фосфорилирования. Мутации в этих белках приводит к нарушениям производства энергии в митохондриях. Этот дефицит клеточной энергии быстрее всего проявляется в тканях, которые сильно зависят от аэробного метаболизма, таких как мозг, скелетные и сердечные мышцы, органы чувств и почки. Это лишь один фактор, участвующий в представлении митохондриальных заболеваний.
Есть и другие факторы, влияющие на проявление митохондриальной болезни, кроме величины и расположения мутации. Митохондрии дублируются посредством деления клеток во время беременности и в течение всей жизни. Поскольку мутация митохондриальной болезни чаще всего встречается на ранних сроках беременности при этих заболеваниях, только митохондрии в мутированной линии являются дефектными. Это приводит к неравномерному распределению дисфункциональных митохондрий в каждой клетке, и в различных тканях тела. Это называется гетероплазмия, которая характерна для митохондриальных заболеваний, включая KSS. Распределение мутантных мтДНК в каждой клетке, ткани и органе, зависит от того, когда и где произошла мутация[16]. Это может объяснить, почему два пациента с одинаковыми мутациями в мтДНК могут представлять совершенно разные фенотипы и, в свою очередь различные синдромы. Публикация в 1992 году Fischel-Ghodsian и др. определила удаление одного и того же 4977-б.п. в мтДНК у двух пациентов с двумя совершенно различными заболеваниями. Один из пациентов имел характерный KSS, а другой пациент — совсем другое заболевание, известное как синдром Пирсона[17]. Усложняет дело то обстоятельство, что в некоторых случаях синдром Пирсона, как было выявлено, может прогрессировать внутри KSS позднее в жизни[18].
Более поздние исследования привели к выводу, что дублирование мтДНК может также играть важную роль в определении присутствия фенотипа. Дублирование мтДНК, кажется, характерно для всех случаев KSS и синдрома Пирсона, в то время как они отсутствуют в CPEO[18][19].
Удаления мтДНК в KSS различаются по размеру (1.3-8kb), а также положению в митохондриальном геноме. Наиболее распространенным удалением является 4.9kb и простирается от позиции 8469 до позиции 13147 в геноме. Это удаление присутствует примерно у ⅓ людей с KSS.
Диагностика
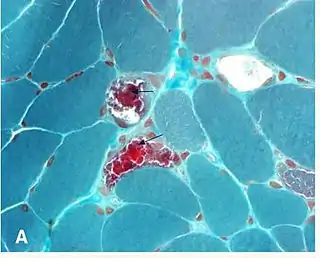
Нейроофтальмологи, как правило, участвуют в диагностике и лечении KSS. Человек должен быть заподозрен в КСС на основе клинических данных экспертизы. Подозрение на миопатию должно быть усилено для пациентов с отсутствием офтальмоплегии и определенного набора параличей черепных нервов (паралич глазодвигательного нерва, паралич блокового нерва, паралич отводящего нерва). Первоначально исследования изображения часто проводится, чтобы исключить более общие патологии. Диагноз может быть подтвержден мышечной биопсией, которая может быть дополнена PCR для определения мутаций мтДНК. Биопсия: Это не обязательно биопсия глазной мышцы, для демонстрации гистопатологические аномалий. Сечение мышечных волокон, трихромное окрашение пятна Гомори рассматривается с помощью световой микроскопии. В мышечных волокнах, с более высоким содержанием мутированных митохондрий, существует более высокая концентрация митохондрий. Это придаёт этим волокнам темно-красный цвет, в результате чего общий вид биопсии описывается как «рваные красные волокна». Аномалии могут быть также продемонстрированы в образцах биопсии мышц с использованием других гистохимических исследований, таких как пятен митохондриальных ферментов, с помощью электронной микроскопии, биохимических анализов мышечной ткани (то есть активность ферментной цепи переноса электронов) и с помощью анализа мышечной митохондриальной ДНК[20].
Лабораторные исследования
Уровень молочной кислоты и пировиноградной кислоты крови, как правило, повышается в результате увеличения анаэробного метаболизма и уменьшением соотношения АТФ/АДФ. При исследовании СМЖ обнаруживается повышенный уровень белка, как правило, > 100 мг/дл, а также повышенный уровень молочной кислоты.[10]
Ведение и лечение
В настоящее время нет медицинского лечения для KSS. Потому что это редкое состояние, есть только сообщения о случаях лечения с очень небольшим количеством данных, чтобы говорить об их эффективности. Было зарегистрировано несколько перспективных открытий, которые могут поддержать открытие новых методов лечения при дальнейших исследованиях. Спутниковые клетки отвечают за регенерацию мышечных волокон. Было отмечено, что мутант мтДНК не обнаруживается или редко обнаруживается в сателлитных клеток, культивируемых у пациентов с KSS. Shoubridge и др. (1997) задался вопросом, может ли первотип мтДНК быть восстановлен в мышечной ткани, при стимуляции регенерации мышц? В вышеупомянутом исследовании, регенерирующие мышечные волокна были отобраны в исходной биопсии, и было обнаружено, что они существенно гомоплазмичны для первотипа мтДНК[16]. Возможно, будущие методы содействия регенерации мышечных клеток и пролиферации спутниковых клеток, позволят значительно улучшить функциональное состояние KSS пациентов.
Одно исследование описывало пациента с KSS, которое позволило сократить уровень сыворотки кофермента Q10. Введение 60-120 мг коэнзима Q10 в течение 3 месяцев привело к нормализации уровней молочной кислоты и пировиноградной кислоты, улучшению ранее диагностированной первой степени AV блока, и совершенствованию глазных движений[21].
Скрининг ЭКГ рекомендуется для всех пациентов с CPEO. При KSS рекомендуется имплантация кардиостимулятора, следить за развитием значительного нарушения сердечной проводимости, даже у бессимптомных пациентов[22].
Скрининг эндокринологических нарушений должен быть выполнен, включая измерение уровня глюкозы в сыворотке крови, функции щитовидной железы, уровней кальция и магния и уровней электролита в сыворотке. Гиперальдостеронизм наблюдается у 3 % пациентов с KSS[23].
Ссылки
- Наблюдение результатов назначения фолиниевой кислоты пациентам с церебральной фолатной недостаточностью и синдромом Кернса-Сейра - русский перевод исследования, осуществленного в 2014 году испанскими учеными.[13]
- Нарушение работы сосудистого сплетения мозга при синдроме Кернса-Сейра - русский перевод заметки в журнале "Cerebrospinal Fluid Research", 2010 год.
Примечания
- Harvey J.N., Barnett D. Endocrine dysfunction in Kearns-Sayre syndrome (неопр.) // Clin. Endocrinol. (Oxf). — 1992. — July (т. 37, № 1). — С. 97—103. — doi:10.1111/j.1365-2265.1992.tb02289.x. — PMID 1424198.
- Kearns T.P., Sayre G.P. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases (англ.) // AMA Arch Ophthalmol : journal. — 1958. — August (vol. 60, no. 2). — P. 280—289. — doi:10.1001/archopht.1958.00940080296016. — PMID 13558799.
- Jager B.V., Fred H.L., Butler R.B., Carnes W.H. Occurrence of retinal pigmentation, ophthalmoplegia, ataxia, deafness and heart block. Report of a case, with findings at autopsy (англ.) // The American Journal of Medicine : journal. — 1960. — November (vol. 29, no. 5). — P. 888—893. — doi:10.1016/0002-9343(60)90122-4. — PMID 13789175.
- Kearns T.P. External Ophthalmoplegia, Pigmentary Degeneration of the Retina, and Cardiomyopathy: A Newly Recognized Syndrome (англ.) // Trans Am Ophthalmol Soc : journal. — 1965. — Vol. 63. — P. 559—625. — PMID 16693635.
- Zeviani M., Moraes C.T., DiMauro S., et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome (англ.) // Neurology : journal. — Wolters Kluwer, 1988. — September (vol. 38, no. 9). — P. 1339—1346. — doi:10.1212/wnl.38.9.1339. — PMID 3412580.
- Lestienne P., Ponsot G. Kearns-Sayre syndrome with muscle mitochondrial DNA deletion (англ.) // The Lancet : journal. — Elsevier, 1988. — April (vol. 1, no. 8590). — P. 885. — doi:10.1016/S0140-6736(88)91632-7. — PMID 2895391.
- Carod-Artal F.J., Lopez Gallardo E., Solano A., Dahmani Y., Herrero M.D., Montoya J. [Mitochondrial DNA deletions in Kearns-Sayre syndrome] (исп.) // Neurologia. — 2006. — Сентябрь (т. 21, № 7). — С. 357—364. — PMID 16977556.
- Lertrit P., Imsumran A., Karnkirawattana P., et al. A unique 3.5-kb deletion of the mitochondrial genome in Thai patients with Kearns-Sayre syndrome (англ.) // Human Genetics : journal. — 1999. — Vol. 105, no. 1—2. — P. 127—131. — doi:10.1007/s004390051074. — PMID 10480366. Архивировано 29 сентября 2000 года.
- Soga F., Ueno S., Yorifuji S. [Deletions of mitochondrial DNA in Kearns-Sayre syndrome] (яп.) // Nippon Rinsho. — 1993. — Сентябрь (т. 51, № 9). — С. 2386—2390. — PMID 8411717.
- Syndrome 950897, раздел Kearns-Sayre Syndrome (англ.) на сайте EMedicine
- Walsh & Hoyt's Clinical Neuro-Ophthalmology: The Essentials (англ.) / Miller, Neil R.; Newman, Nancy J.. — Lippincott Williams & Wilkins, 2007.
- Garcia-Cazorla A., Quadros E.V., Nascimento A., Garcia-Silva M.T., Briones P., Montoya J et al. Mitochondrial diseases associated with cerebral folate deficiency (англ.) // Neurology : journal. — Wolters Kluwer, 2008. — Vol. 70, no. 16. — P. 1360—1362. — doi:10.1212/01.wnl.0000309223.98616.e4. — PMID 18413591.
- Quijada-Fraile P., O'Callaghan M., Martín-Hernández E., Montero R., Garcia-Cazorla À, de Aragón AM et al. Follow-up of folinic acid supplementation for patients with cerebral folate deficiency and Kearns-Sayre syndrome (англ.) // Orphanet Journal of Rare Diseases : journal. — 2014. — Vol. 9. — P. 217. — doi:10.1186/s13023-014-0217-2. — PMID 25539952.
- Spector R., Johanson C.E. Choroid plexus failure in the Kearns-Sayre syndrome (англ.) // Cerebrospinal Fluid Res : journal. — 2010. — Vol. 7. — P. 14. — doi:10.1186/1743-8454-7-14. — PMID 20731822.
- Fine P.E. Mitochondrial inheritance and disease (англ.) // The Lancet. — Elsevier, 1978. — September (vol. 2, no. 8091). — P. 659—662. — doi:10.1016/S0140-6736(78)92764-2. — PMID 80581.
- Shoubridge E.A., Johns T., Karpati G. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy (англ.) // Human Molecular Genetics : journal. — Oxford University Press, 1997. — December (vol. 6, no. 13). — P. 2239—2242. — doi:10.1093/hmg/6.13.2239. — PMID 9361028.
- Fischel-Ghodsian N., Bohlman M.C., Prezant T.R., Graham J.M., Cederbaum S.D., Edwards M.J. Deletion in blood mitochondrial DNA in Kearns-Sayre syndrome (англ.) // Pediatric Research : journal. — 1992. — June (vol. 31, no. 6). — P. 557—560. — doi:10.1203/00006450-199206000-00004. — PMID 1635816.
- Poulton J., Morten K.J., Weber K., Brown G.K., Bindoff L. Are duplications of mitochondrial DNA characteristic of Kearns-Sayre syndrome? (англ.) // Human Molecular Genetics : journal. — Oxford University Press, 1994. — June (vol. 3, no. 6). — P. 947—951. — doi:10.1093/hmg/3.6.947. — PMID 7951243.
- Miller, Neil R.; Newman, Nancy J.; Bioussee, Valerie; Kerrison, John B. Ch. 20, adapted from a chapter 22 by Paul N. Hoffman // Walsh and Hoyt's Clinical Neuro-ophthalmology: the essentials (англ.). — Philadelphia: Lippincott Williams & Wilkins, 2008. — P. 432—436.
- Rubin, Richard M.; Sadun, Alfredo A. Ch. 9.17 Ocular Myopathies // Ophthalmology (неопр.) / Yanoff, Myron; Duker, Jason. — 3rd. — Mosby, 2008.
- Ogasahara S., Yorifuji S., Nishikawa Y., et al. Improvement of abnormal pyruvate metabolism and cardiac conduction defect with coenzyme Q10 in Kearns-Sayre syndrome (англ.) // Neurology : journal. — Wolters Kluwer, 1985. — March (vol. 35, no. 3). — P. 372—377. — doi:10.1212/WNL.35.3.372. — PMID 3974895.
- Gregoratos G., Abrams J., Epstein A.E., et al. ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/NASPE Committee to Update the 1998 Pacemaker Guidelines) (англ.) // Circulation : journal. — Lippincott Williams & Wilkins, 2002. — October (vol. 106, no. 16). — P. 2145—2161. — doi:10.1161/01.CIR.0000035996.46455.09. — PMID 12379588.
- Syndrome 950897, раздел Kearns-Sayre Syndrome (англ.) на сайте EMedicine