Бета-маннозидоз
Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы[3].
| Бета-маннозидоз | |
|---|---|
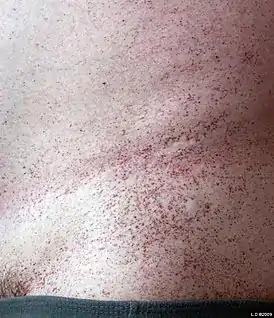 Ангиокератома туловища — клинический признак, ассоциированный с β-маннозидозом[1] | |
| МКБ-10 | E77.1 |
| МКБ-10-КМ | E77.1 |
| МКБ-9 | 271 |
| МКБ-9-КМ | 271.8[2] |
| OMIM | 248510 |
| DiseasesDB | 34529 |
| MeSH | D044905 |
Патогенез
Генетически детерминированный дефект фермента β-маннозидазы, участвующего в расщеплении сложных углеводов в лизосомах ведёт к накоплению метаболитов внутри клетки с последующим нарушением её функции. Этот фермент кодируется геном MANBA, который расположен на длинном плече 4-й хромосомы (локус 4q22-25).
Наследование

Маннозидоз наследуется, как и подавляющее большинство лизосомных болезней накопления, по аутосомно-рецессивному типу наследования[3][4]. Следовательно, с одинаковой частотой встречается как у мужчин, так и у женщин. Заболевание клинически манифестирует только в случае, когда обе аутосомы, полученные по одной от отца и матери, являются дефектными (повреждение обеих копий гена MANBA, находящихся на гомологичных аутосомах, локус 4q22-25).
Клиническая картина
Новорождённые пациенты кажутся здоровыми, а развитие болезни отличается разнообразными клиническими проявлениями. Начало инфантильной формы заболевания характеризуется наличием более выраженных проявлений нейродегенерации, вплоть до развития интеллектуальной инвалидности у некоторых детей. Потеря слуха и наличие ангиокератом[1] — наиболее частые признаки заболевания. Однако, в связи с тем, что данная генетическая патология встречается крайне редко, полный фенотип болезни до конца не изучен[5].
В результате своей редкости и не специфичности клинических проявлений, диагноз: β-маннозидоз может быть установлен с опозданием. Описаны случаи ретроспективной диагностики у взрослых пациентов на фоне сформировавшейся интеллектуальной инвалидности, сопровождающейся расстройствами поведения, включая агрессию[6].
См. также
Примечания
- Rodriguez-Serna, M., Botella-Estrada, R., Chabas, A., Coll, M.-J., Oliver, V., Febrer, M.-I., Aliaga, A.. Angiokeratoma corporis diffusum associated with beta-mannosidase deficiency. Arch. Derm. 132: 1219-1222, 1996. PubMed: 8859034. ncbi.nlm.nih.gov. Дата обращения: 23 января 2015.
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- OMIM 248510
- Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., Медицина, 1996, 320 с.: ил. Глава 316. Лизосомные болезни накопления (с. 250—273). med-books.info. Дата обращения: 23 января 2015.
- Enns, Gregory M.; Steiner, Robert D.; Cowan, Tina M. Lysosomal Disorders // Pediatric Endocrinology and Inborn Errors of Metabolism (англ.) / Sarafoglou, Kiriakie; Hoffmann, Georg F.; Roth, Karl S.. — 1st. — New York: McGraw-Hill Medical, 2009. — P. 721—755. — ISBN 978-0-07-143915-2.
- Sedel F., Baumann N., Turpin J. C., Lyon-Caen O., Saudubray J. M., Cohen D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. (англ.) // Journal of inherited metabolic disease. — 2007. — Vol. 30, no. 5. — P. 631—641. — doi:10.1007/s10545-007-0661-4. — PMID 17694356.