Эпигеномика
Эпигено́мика (англ. Epigenomics) — раздел молекулярной биологии, изучающий совокупность эпигенетических модификаций генетического материала клетки (эпигеном) с помощью высокопроизводительных методов. Эпигеномика аналогична геномике и протеомике, которые изучают геном и протеом клетки, соответственно[1].
Эпигенетические модификации — это обратимые ковалентные химические модификации клеточной ДНК и гистонов, которые влияют на экспрессию генов, не изменяя нуклеотидную последовательность ДНК[2]. Механизмы внесения ковалентных модификаций ДНК и гистонов и их влияния на экспрессию генов являются предметом изучения эпигенетики, а эпигеномика изучает результат — эпигенетические модификации и их распределение по геному при помощи молекулярных методов, таких как ChIP-seq, бисульфитное секвенирование и другие.
Самые изученные эпигенетические модификации — это метилирование ДНК и модификации гистонов. Изучение эпигенома на глобальном уровне стало возможно лишь относительно недавно в связи с развитием высокопроизводительных методов изучения генома[3][4].
Эпигенетические модификации
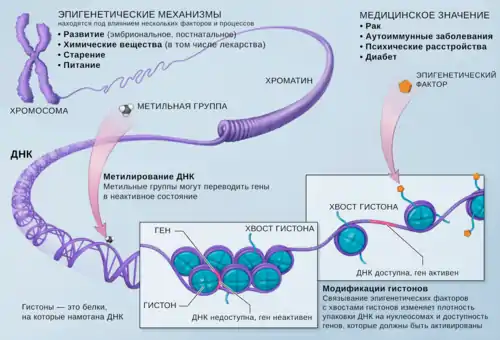
Модификации генома, которые изменяют экспрессию генов, но не связаны с изменением первичной последовательности ДНК (нуклеотидной последовательности) и передаются при митотическом и мейотическом делении клетки, называют эпигенетическими модификациями. Наиболее изучены такие эпигенетические модификации, как метилирование ДНК и модификации гистонов[2]. Совокупность всех эпигенетических модификаций ДНК в клетке называют эпигеном. Клетка поддерживает свой эпигеном на протяжении всей жизни, поскольку стабильность эпигенома непосредственно связана со стабильностью генома, так как эпигеном вовлечён в важнейшие клеточные процессы, например, репарацию ДНК[5][6]. Изменения эпигенома клетки могут привести к превращению её в злокачественную[7][4][8]. Изменения эпигенома могут быть связаны с развитием и других заболеваний человека, помимо разных видов рака, таких как сахарный диабет 2-го типа, болезнь Альцгеймера, болезнь Паркинсона, синдром ломкой X-хромосомы и синдром Прадера — Вилли[9].
Метилирование ДНК
Первой открытой эпигенетической модификацией стало метилирование ДНК. Метилирование ДНК заключается в присоединении метильной группы к нуклеотидам в составе ДНК. Ферменты, катализирующие эту реакцию, известны как ДНК-метилтрансферазы. Хотя метилирование ДНК — это стабильная и наследуемая модификация, существуют ферменты, удаляющие метильные метки с ДНК (ДНК-деметилазы). У эукариот метильная группа чаще всего присоединяется к пятому атому углерода азотистого основания цитозина в составе ДНК с образованием 5-метилцитозина, или 5mC (как правило, в составе CpG-островков, в которых остатки цитозина соседствуют с остатками гуанина)[3][10].
Нуклеотидные позиции, подвергающиеся метилированию, значительно различаются у разных видов и даже среди клеток одного организма. Животные используют метилирование ДНК по-разному; так, уровень метилирования ДНК у позвоночных очень высок, а у беспозвоночных он имеет средние значения. У некоторых организмов, таких как нематода Caenorhabditis elegans, 5mC и ДНК-метилтрансферазы полностью отсутствуют. Вероятно, в этих случаях роль метилирования ДНК выполняют иные механизмы[11].
В пределах одного организма уровень метилирования ДНК может значительно различаться на разных стадиях развития и в зависимости от участка генома. Например, у мыши примордиальные клетки зародышевой линии претерпевают полногеномное деметилирование, но на стадии имплантации зародыша метильные метки возвращаются на позиции, в которых они присутствовали у соматических клеток[11]. Когда метилирование ДНК затрагивает промоторную область, то транскрипция соответствующего гена подавляется. Напротив, активно экспрессируемые гены, как правило, имеют неметилированные промоторы[3].
Механизм репрессии экспрессии генов через метилирование ДНК включает несколько этапов. С метилированными и неметилированными остатками цитозина взаимодействуют разные ДНК-связывающие белки. Через них к участкам с 5mC привлекаются гистондеацетилазы, которые запускают ремоделирование хроматина, в результате которого ДНК становится недоступной для взаимодействия с компонентами аппарата транскрипции, такими как РНК-полимераза, что приводит к эффективной репрессии экспрессии генов[12].
Модификации гистонов
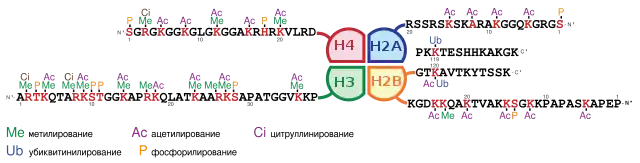
У эукариот геномная ДНК сопряжена с белками, образуя хроматин. Самые многочисленные белки хроматина — гистоны, на которые намотана двойная спираль ДНК. Гистоны обогащены положительно заряженными аминокислотами, что облегчает их связывание с отрицательно заряженным сахарофосфатным остовом ДНК за счёт электростатических взаимодействий. Основные повторяющиеся структурные единицы хроматина — нуклеосомы — представляют собой октамер, включающий по две молекулы коровых гистонов H2A, H2B, H3 и H4, на который намотана нить ДНК длиной 146 пар оснований (п. о.). Нуклеосомы и намотанная на них ДНК формируют хроматиновую фибриллу диаметром 10 нм, которая может подвергаться дальнейшей конденсации[13][14].
Степень компактизации хроматина зависит от стадии клеточного цикла и может быть различной в разных участках генома[15]. Степень конденсации хроматина связана с его транскрипционной активностью. Неконденсированный хроматин более транскрипционно-активен, чем плотно упакованный хроматин, так как он более доступен для аппарата транскрипции. Поэтому экспрессию гену можно модулировать за счёт ремоделирования хроматина и регулирования плотности его упаковки[14].
Ремоделирование хроматина осуществляется при помощи внесения посттрансляционных модификаций на N-концевые хвосты коровых гистонов[16]. Совокупность модификаций гистонов в данной клетке называется гистоновым кодом. Известно множество видов модификаций гистонов: ацетилирование, метилирование, фосфорилирование, убиквитинилирование, SUMOилирование, АДФ-рибозилирование, дезаминирование и изомеризация пролина. Ацетилирование, метилирование, фосфорилирование и убиквитинилирование часто связаны с активацией экспрессии генов, а метилирование, убиквитинилирование, SUMOилирование, дезаминирование и изомеризация пролина могут приводить к репрессии гена. Стоит отметить, что некоторые модификации, такие как метилирование, фосфорилирование и убиквинитилирование могут влиять на транскрипционную активность генов в зависимости от специфических аминокислотных остатков модифицируемых геномов. Кроме того, свой вклад вносит и модифицируемый участок генома. Так, метилирование лизина 36 гистона H3 (H3K36me) в кодирующей области гена приводит к его активации, а в промоторе — напротив, к инактивации[14].
Модификации гистонов влияют на экспрессию генов при помощи двух основных механизмов: за счёт разрушения контактов между нуклеосомами и за счёт привлечения АТФаз, ремоделирующих хроматин. Первый механизм реализуется при ацетилировании остатков лизина на хвостах гистонов, которое катализируется гистонацетилтрансферазами. Гистонацетилтрансферазы входят в состав мультибелковых комплексов, которые привлекаются к хроматину при связывании с ДНК белков-активаторов. Ацетилирование нивелирует положительный заряд остатка лизина, который стабилизирует взаимодействие нуклеосом с отрицательно заряженным остовом ДНК. Ацетилированные остатки лизина в составе гистонов способствуют диссоциации нуклеосом и декомпактизации хроматина. В декомпактизованном хроматине ДНК более доступна для аппарата транскрипции, поэтому ген становится более активным. Деацетилазы могут удалять ацетильные группы с хвостов гистонов[14][16].
Второй механизм включает привлечение комплексов ремоделирования хроматина через связывание белков-активаторов с соответствующими энхансерами. Комплексы ремоделирования хроматина меняют положение нуклеосом несколькими способами, что может как повышать, так и понижать доступность ДНК для аппарата транскрипции. Примером комплекса ремоделирования хроматина может служить комплекс SWI/SNF дрожжей; он регулирует экспрессию нескольких генов через перестройки хроматина[14][17].
Методы
Целью эпигеномики является поиск и описание эпигенетических меток на глобальном уровне, подобно тому, как геномика изучает всю совокупность генетического материала клетки, а протеомика — совокупность всех клеточных белков[1]. Подобно геномике и протеомике, важнейшую методологическую часть эпигеномики составляют биоинформатические подходы[18].
Анализ модификаций гистонов
Процессы транскрипции, репликации и репарации ДНК требуют взаимодействия геномной ДНК с ядерными белками. Известно, что некоторые участки генома особенно чувствительны к действию ДНКазы I, которая неспецифично расщепляет ДНК, не защищённую белками. Такие гиперчувствительные сайты считались транскрипционно активными участками генома, что подтвердилось их ассоциацией с РНК-полимеразой, а также топоизомеразами I и II[19]. В гиперчувствительных сайтах плотность упаковки ДНК понижена. Чаще всего гиперчувствительные сайты соответствуют промоторам, в которых ДНК должна быть открытой, чтобы с ней могли связаться компоненты аппарата транскрипции[20].
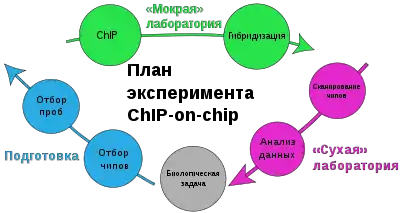
Полногеномный поиск эпигенетических модификаций впервые был выполнен с помощью метода иммунопреципитации хроматина (англ. Chromatin immunoprecipitation, ChIP) вместе с ДНК-микрочипами (эта технология известна как ChIP-on-chip)[13]. В случае ChIP-on-chip с помощью иммунопреципитации хроматина выделяют не транскрипционные факторы и ДНК-связывающие белки-активаторы, а модифицированные гистоны. Сначала гистоны in vivo ковалентно сшивают с ДНК при помощи обработки клеток формальдегидом. Далее клетки подвергают лизису, что делает возможным экстракцию и фрагментацию хроматина. Фрагментацию хроматина проводят с помощью обработки ультразвуком или эндонуклеазой рестрикции. В ходе такой обработки участки хроматина, не связанные с белками, разрушаются, так что остаются только комплексы ДНК с белками. Далее с помощью антител, специфичных к гистонам с определёнными модификациями, осуществляют иммунопреципитацию комплексов ДНК с такими гистонами[14]. После иммунопреципитации ДНК и гистоны разделяют, фрагменты ДНК амплифицируют с помощью полимеразной цепной реакции и метят флуоресцентной меткой (например, Cy3 или Cy5). На финальной стадии флуоресцентно-меченную ДНК гибридизуют с фрагментами геномной ДНК, иммобилизованными на ДНК-микрочипе. По интенсивности сигнала, соответствующего каждому фрагменту, делают вывод о том, с какими из них взаимодействуют гистоны с интересующей эпигенетической меткой[21][22].
С помощью ChIP-on-chip был изучен эпигеном пекарских дрожжей, на основании этого были сделаны выводы о функциях определённых модификаций гистонов. Было показано, с какими эпигенетическими метками связаны репрессия или активация транскрипции и в каких участках генома они происходят. Хотя с помощью ChIP-on-chip удалось изучить эпигеном дрожжей с почти полным покрытием, применение этого метода для организмов с более крупными геномами, таких как человек, ограничено[13][14].
.png.webp)
Для полногеномного изучения эпигенетических меток на больших геномах вместе с иммунопреципитацией хроматина используют высокопрозводительные методы, такие как SAGE (от англ. Serial Analysis of Gene Expression), секвенирование спаренных концов (PET от англ. Paired-end tag) и высокопроизводительное секвенирование (этот метод известен как ChIP-seq). ChIP-seq включает стандартный протокол иммунопреципитации хроматина, однако вместо амплификации выделенной ДНК и её гибридизации на микрочипе её секвенируют с помощью высокопроизводительных методов. ChIP-seq показал себя как эффективный метод для анализа модификаций гистонов на глобальном уровне, выявления сайтов связывания белков, взаимодействующих с ДНК, причём с большим разрешением, чем дают другие методы[13][21].
Анализ метилирования ДНК
Методы определения нуклеотидной последовательности ДНК не подходят для обнаружения в ней метилированных нуклеотидов. В частности, при проведении полимеразной цепной реакции (ПЦР) или бактериального клонирования метильные метки не сохраняются при удвоении ДНК, и эпигенетическая информация теряется. Методы гибридизации ДНК, в которых для определения последовательности исследуемых фрагментов ДНК и их положения в геноме используются радиоактивные пробы, также не подходят, поскольку они не различают метилированную и неметилированную ДНК[23][3].
Первый разработанный метод детекции метилированных нуклеотидов основан на применении эндонуклеаз рестрикции (рестриктаз). В этом методе геномную ДНК обрабатывают двумя рестриктазами, которые распознают одну и ту же последовательность, но одна из них чувствительна к метилированию, а вторая — нет. Идея метода заключается в том, что метилированный сайт может быть распознан и разрезан только рестриктазой, не чувствительной к метилированию. Сравнивая фрагменты, полученные при обработке ДНК рестриктазой, чувствительной к метилированию и не чувствительной к нему, можно выявить участки, подвергающиеся метилированию. Этот этап включает амплификацию фрагментов, полученных в результате обработки рестриктазами, с помощью ПЦР, разделение их с помощью гель-электрофореза и анализ с помощью саузерн-блота[23][3].
Описанный выше метод был использован для анализа метилирования ДНК в локусе гемоглобинов человека. Различные гены этого локуса (γ-, δ- и β-глобины) экспрессируются на разных стадиях развития организма[24]. Исследование подтвердило, что те гены, которые не экспрессировались, были обогащены 5mC[25].
Метод, основанный на применении рестриктаз, не позволяет изучить метилирование на уровне целого генома, то есть метилом. Даже в пределах локуса он не даёт полного представления о количестве и расположении метильных меток, так как информацию о метилировании можно получить только с тех участков, которые содержат сайты узнавания используемых рестриктаз. В связи с этим метод даёт большое количество ложноотрицательных результатов[3].
Впервые метилирование на уровне всего генома было изучено с помощью метода, известного как Restriction landmark genomic scanning (RLGS, дословно «сканирование генома на предмет сайтов рестрикции»). В этом методе также используются ферменты, расщепляющие ДНК и чувствительные к её метилированию, однако разделение фрагментов происходит в ходе двумерного гель-электрофореза, что позволяет изучить расположение метильных меток детальнее[3].
Однако получить полное представление о метилировании геномной ДНК с высоким разрешением стало возможным только с появлением ДНК-микрочипов и методов секвенирования нового поколения[26]. Как и в RLGS, новые подходы включают расщепление ДНК эндонуклеазами. В случае гибридизации с дифференциальным метилированием (англ. differential methylation hybridization, DMH) одна проба геномной ДНК обрабатывается рестриктазами, чувствительными к метилированию, а другая — не чувствительными к метилированию ферментами. Далее фрагменты обеих проб амплифицируют и метят различными флуоресцентными метками, после чего наносят фрагменты из обеих проб на один микрочип. Уровень метилирования ДНК в данном локусе определяют как разность относительной интенсивности свечения двух красителей. Использование высокопроизводительного секвенирования позволяет добиться большего разрешения, чем применение ДНК-микрочипов. Микрочип для данного метода должен быть отнормирован в соответствии с плотностью CpG-островков, чтобы давать достоверные результаты. В частности, с помощью секвенирования можно выявить аллель-специфичное метилирование, кроме того, этот подход позволяет работать с более крупными геномами и не требует создания отнормированных микрочипов[3].
Бисульфитное секвенирование включает химическое превращение неметилированных остатков цитозина, поэтому далее их удаётся выявить с помощью обычного секвенирования. При обработке бисульфитом натрия и щёлочью неметилированный цитозин превращается в урацил, а метилированный цитозин остаётся интактным. Дальнейшая амплификация и секвенирование ДНК, не обработанной бисульфитом натрия, и ДНК, подвергшейся обработке, позволяет идентифицировать сайты метилирования. Подобно классическим методам, основанным на применении чувствительных к метилированию рестриктаз, бисульфитное секвенирование сначала применяли для изучения метилирования в пределах отдельных локусов, однако появление методов полногеномного секвенирования позволило использовать его для анализа метилирования на уровне целого генома. Однако в отличие от методов, основанных на применении рестриктаз, бисульфитное секвенирование даёт возможность выявить сайт метилирования с точностью до одного нуклеотида. К числу ограничений бисульфитного метода можно отнести неполное превращение метилированного цитозина в урацил, которое приводит к появлению ложноположительных результатов. Кроме того, при бисульфитном секвенировании может происходить разрушение ДНК, и протокол метода включает стадию удаления бисульфита натрия[23][3].
Для полногеномного анализа метилирования в паре с бисульфитным секвенированием используют технологии секвенирования нового поколения. Сочетание этих методов позволяет установить сайты метилирования с наибольшим метилированием из возможных, однако на этапе сборки прочтений в геном возникает много сложностей, связанных с пониженной сложностью последовательности ДНК, обработанной бисульфитом. Для борьбы с этим эффектом можно увеличивать длину прочтений; на таком подходе основано полногеномное секвенирование методом дробовика (англ. whole genome shotgun bisulphite sequencing, WGBS). Метод WGBS, использующий платформу Illumina Genome Analyzer, уже был применён для анализа метилома растения Arabidopsis thaliana[3]. Существуют также варианты бисульфитного секвенирования с ограниченной представленностью[27][28], которые особенно актуальны в случае организмов с большими геномами[29].
Анализ доступности хроматина
Под доступностью хроматина понимают меру того, насколько соответствующий ему участок генома открыт для взаимодействия с транскрипционными факторами и другими компонентами аппарата транскрипции. Недоступные участки, плотно упакованные с помощью нуклеосом, не подвергаются активной транскрипции, в отличие от открытых участков[30]. Изменение доступности хроматина — это важный эпигенетический регуляторный процесс, благодаря которому достигается контекстоспецифичный или клеткоспецифичный уровень экспрессии определённых генов[31]. Для изучения доступности хроматина в клетке применяются различные методы, такие как MNase-seq, DNase-seq, ATAC-seq и FAIRE-seq. Ключевой особенностью этих подходов является их способность к отделению ДНК, связанной с гистонами, от свободной ДНК. Эти последовательности затем выравниваются на референсный геном, что позволяет установить их расположение[32].
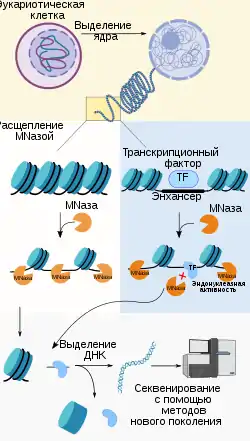
MNase-seq и DNase-seq основаны на одном и том же принципе, а именно, расщеплении нуклеазами свободной ДНК, не связанной с гистонами или другими белками. В то же время фрагменты ДНК, взаимодействующие с белками, остаются нетронутыми, так что их можно отделить от белков и анализировать далее. Поскольку эти методы предполагают разрушение активных участков генома, их позицию можно установить лишь косвенно с помощью секвенирования уцелевших фрагментов ДНК и их выравнивания на референсный геном. В методе MNase-seq используется микрококковая нуклеаза, которая вносит одноцепочечный разрыв в цепь, комплементарную последовательности-мишени[33]. В методе DNase-seq используется ДНКаза I, которая неспецифично вносит двуцепочечные разрывы в ДНК[34]. DNase-seq получил такое широкое распространение, что свободные от нуклеосом участки стали называть DHSs (от англ. Nase I hypersensitive sites)[35], а консорциум ENCODE выбрал этот метод для полногеномного анализа доступности хроматина[36]. Главная проблема DNase-seq заключается в том, что разрывы могут вноситься неслучайно, что снижает качество получаемых результатов[37].
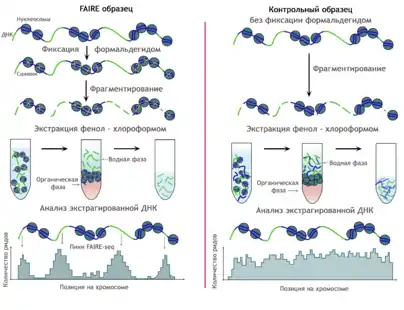
Метод FAIRE-seq (англ. Formaldehyde-Assisted Isolation of Regulatory Elements) начинается с сшивания ДНК с нуклеосомами и последующего расщепления ДНК с помощью ультразвука. Свободные и связанные фрагменты выделяют с помощью стандартной фенол-хлороформной экстракции, при этом фракция белков образует вязкую интерфазу, а свободная ДНК оказывается в водной фазе, откуда её можно взять для дальнейшего анализа[38]. Обработка ультразвуком вносит случайные разрывы, поэтому фактор неслучайности здесь полностью исключён, а фрагменты, получающиеся после воздействия ультразвука, имеют большую длину, чем при ферментативной обработке (200—700 п. о.)[32]. Благодаря этому FAIRE-seq может быть использован для анализа больших участков генома, однако он не даёт разрешения в одну нуклеосому. В отличие от методов, использующих нуклеазы, FAIRE-seq позволяет идентифицировать активные участки генома напрямую и включает более простую стадию пробоподготовки[39].
Метод ATAC-seq основан на применении транспозазы Tn5. Транспозаза вставляет в геном адаптеры для секвенирования, причём с большей частотой в те участки генома, которые свободны от белков. Вставленные транспозазой адаптеры далее используются в ПЦР и последующем секвенировании[40].
Прямое выявление эпигенетических меток
Чувствительность ДНК-полимеразы, использующейся при одномолекулярном секвенировании в реальном времени, делает возможным непосредственно выявлять эпигенетические метки, такие как метильные группы, по мере перемещения полимеразы по секвенируемой молекуле ДНК[41]. В нескольких проектах была показана применимость этого подхода для получения полногеномной эпигенетической информации у бактерий[42][43][44][45].
Нанопоровое секвенирование основано на выявлении изменений силы электрического тока при встрече с модифицированными нуклеотидами (например, метилированными). ДНК-полимераза обеспечивает погрузку одноцепочечной ДНК в пору миниатюрной камеры, заполненной раствором электролита, в которой из-за приложенного напряжения возникает электрический ток. Прохождение определённых нуклеотидов через пору уменьшает её сечение, доступное для ионов электролита, что приводит к уменьшению силы тока[46]. Фиксируя такие изменения силы тока, можно выявить CpG-островки в ДНК, пропускаемой через пору. С помощью нанопорового секвенирования можно даже различить гидроксиметилирование и метилирование. Секвенатор MinION, разработанный Oxford Nanopore Technologies, позволяет отличить неметилированный цитозин от метилированного без предварительной химической обработки[47]. Технология нанопорового секвенирования также реализована в приборах Nanopolish и SignaAlign: первый позволяет оценить частоту метилирования в прочтениях, а второй — его вероятность[48].
Одномолекулярное секвенирование в реальном времени включает использование нулевого световода (англ. Zero-mode waveguide). С нижней частью световода связана единственная молекула ДНК-полимеразы, матрицей для которой служит также единственная молекула ДНК. Каждый из четырёх нуклеотидов ДНК метят одним из четырёх различных флуоресцентных красителей. Когда ДНК-полимераза присоединяет очередной нуклеотид, с него удаляется флуоресцентная метка, а её световой сигнал регистрируется детектором. Когда в ходе секвенирования ДНК-полимераза встречается с модифицированным нуклеотидом в матрице, её кинетика изменяется: она ускоряется или замедляется уникальным для данной модификации способом. В ходе секвенирования флуоресцентные вспышки оцениваются не только по их спектрам эмиссии, но также по длительности и интервалам между вспышками. Эти параметры, известные как ширина пульса и межпульсовый интервал, позволяют оценить кинетику ДНК-полимеразы в данный момент времени. Ширина пульса есть функция всех кинетических этапов от присоединения нуклеотида до высвобождения флуорофора, а межпульсовый интервал определяется кинетикой связывания нуклеотидов и перемещением ДНК-полимеразы. В 2010 году было показано, что одномолекулярное секвенирование в реальном времени позволяет выявить такие модифицированные нуклеотиды, как N6-метиладенозин, 5-метилцитозин и 5-гидроксилцитозин. Эти модификации по-разному изменяют кинетику ДНК-полимеразы, что и позволяет отличать их друг от друга[49].
В 2017 году был предложен комбинированный метод, включающий бисульфитное превращение неметилированных остатков цитозина и одномолекулярное секвенирование в реальном времени. Этот подход известен как одномолекулярное бисульфитное секвенирование в реальном времени (англ. single-molecule real-time bisulfite sequencing, SMRT-BS) и представляет собой метод для прицельного анализа метилирования CpG-островков[50].
Теоретические модели
Первые математические модели для различных состояний нуклеосом, влияющих на экспрессию генов, были предложены в 1980-х годах. В дальнейшем эти идеи были почти полностью забыты, пока не появились экспериментальные данные о ковалентных модификациях гистонов и гистоновом коде[51]. В последующем высокопроизводительные методы показали широкое распространение эпигенетических модификаций, что способствовало разработке новых теоретических моделей, описывающих появление, поддержание и изменение этих меток[52]. Большинство из предложенных моделей описывают эпигенетические модификации в терминах одномерной решётки[53].
Редактирование эпигенома
Существует множество исследовательских методов, направленных на редактирование эпигенома. Изменения в эпигеноме происходят и при классических генетических экспериментах, приводящих к нокауту или нокдауну гена-мишени, делеции доменов белка, появлению точечных мутаций, мутаций приобретения или утраты функции. На эпигеном влияет и введение в геном искусственных последовательностей с индуцибельной экспрессией, а также введение в клетку эктопически экспрессируемых векторов. Эпигеном может изменяться под действием биологически активных малых молекул, ингибирующих ферменты, которые отвечают за включение или удаление эпигенетических меток. Примером могут служить азацитидин и децитабин, необратимо ингибирующие ДНК-метилтрансферазы 1 и 3, а также ромидепсин, ингибирующий гистондеацетилазы. Направленное редактирование эпигенома связано с применением методов направленного редактирования генома, в которых используются нуклеазы, содержащие цинковые пальцы, нуклеазы TALEN, а также система CRISPR/Cas9. Редактирующий эффект связан с воздействием на ферменты, которые прямо или опосредованно вовлечены в удаление или внесение эпигенетических меток[9].
История
Термин «эпигенетика» впервые употребил Конрад Хэл Уоддингтон в 1942 году для обозначения молекулярных механизмов, которые определяют фенотипическое проявление гена. В течение последующих 50 лет эти механизмы были тщательно изучены, а также показано их влияние в обеспечении пластичности отдельных клеток и организма в целом[54].
Термин «эпигеномика» был предложен в 1997 году[55], когда благодаря развитию высокопроизводительных методов стало возможным изучение эпигенетических модификаций на уровне всего генома. В 2000-х началась работа по составлению карты эпигенома человека (англ. Human Epigenome Project)[56]. В 2003 году начал работу проект ENCODE, который стал первым международным проектом, использовавшим эпигеномные данные для поиска регуляторных элементов в геноме человека. В 2010 году был основан Консорциум по эпигеному человека (англ. International Human Epigenome Consortium, IHEC), целью которого является получение 1000 референсных эпигеномов клеток различных типов[54].
Примечания
- Russell, 2010, p. 217, 230.
- Russell, 2010, p. 475.
- Laird P. W. Principles and challenges of genomewide DNA methylation analysis. (англ.) // Nature Reviews. Genetics. — 2010. — March (vol. 11, no. 3). — P. 191—203. — doi:10.1038/nrg2732. — PMID 20125086.
- Zhu J., Adli M., Zou J. Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P. L., Bennett D. A., Houmard J. A., Muoio D. M., Onder T. T., Camahort R., Cowan C. A., Meissner A., Epstein C. B., Shoresh N., Bernstein B. E. Genome-wide chromatin state transitions associated with developmental and environmental cues. (англ.) // Cell. — 2013. — 31 January (vol. 152, no. 3). — P. 642—654. — doi:10.1016/j.cell.2012.12.033. — PMID 23333102.
- Alabert C., Groth A. Chromatin replication and epigenome maintenance. (англ.) // Nature Reviews. Molecular Cell Biology. — 2012. — 23 February (vol. 13, no. 3). — P. 153—167. — doi:10.1038/nrm3288. — PMID 22358331.
- Ghosh S., Sinha J. K., Raghunath M. Epigenomic maintenance through dietary intervention can facilitate DNA repair process to slow down the progress of premature aging. (англ.) // IUBMB Life. — 2016. — September (vol. 68, no. 9). — P. 717—721. — doi:10.1002/iub.1532. — PMID 27364681.
- The Potential Epigenetic and Anticancer Power of Dietary Flavones (11 октября 2016).
- Russell, 2010, p. 597.
- Holtzman L., Gersbach C. A. Editing the Epigenome: Reshaping the Genomic Landscape. (англ.) // Annual Review Of Genomics And Human Genetics. — 2018. — 31 August (vol. 19). — P. 43—71. — doi:10.1146/annurev-genom-083117-021632. — PMID 29852072.
- Russell, 2010, p. 531—532.
- Bird A. DNA methylation patterns and epigenetic memory. (англ.) // Genes & Development. — 2002. — 1 January (vol. 16, no. 1). — P. 6—21. — doi:10.1101/gad.947102. — PMID 11782440.
- Russell, 2010, p. 532—533.
- Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. (англ.) // Cell. — 2007. — Vol. 129, no. 4. — P. 823—837. — doi:10.1016/j.cell.2007.05.009. — PMID 17512414.
- Kouzarides T. Chromatin modifications and their function. (англ.) // Cell. — 2007. — 23 February (vol. 128, no. 4). — P. 693—705. — doi:10.1016/j.cell.2007.02.005. — PMID 17320507.
- Russell, 2010, p. 24—27.
- Russell, 2010, p. 529—530.
- Russell, 2010, p. 530.
- Russell, 2010, p. 218.
- Gross D. S., Garrard W. T. Nuclease hypersensitive sites in chromatin. (англ.) // Annual Review Of Biochemistry. — 1988. — Vol. 57. — P. 159—197. — doi:10.1146/annurev.bi.57.070188.001111. — PMID 3052270.
- Russell, 2010, p. 529.
- Gibson, Muse, 2009, p. 229—232.
- Russell, 2010, p. 532.
- Eads C. A., Danenberg K. D., Kawakami K., Saltz L. B., Blake C., Shibata D., Danenberg P. V., Laird P. W. MethyLight: a high-throughput assay to measure DNA methylation. (англ.) // Nucleic acids research. — 2000. — Vol. 28, no. 8. — P. 32. — PMID 10734209.
- Russell, 2010, p. 552—553.
- van der Ploeg L. H., Flavell R. A. DNA methylation in the human gamma delta beta-globin locus in erythroid and nonerythroid tissues. (англ.) // Cell. — 1980. — April (vol. 19, no. 4). — P. 947—958. — doi:10.1016/0092-8674(80)90086-0. — PMID 6247075.
- Johannes F., Colot V., Jansen R. C. Epigenome dynamics: a quantitative genetics perspective. (англ.) // Nature Reviews. Genetics. — 2008. — November (vol. 9, no. 11). — P. 883—890. — doi:10.1038/nrg2467. — PMID 18927581.
- Trucchi E., Mazzarella A. B., Gilfillan G. D., Lorenzo M. T., Schönswetter P., Paun O. BsRADseq: screening DNA methylation in natural populations of non-model species. (англ.) // Molecular Ecology. — 2016. — April (vol. 25, no. 8). — P. 1697—1713. — doi:10.1111/mec.13550. — PMID 26818626.
- van Gurp T. P., Wagemaker N. C., Wouters B., Vergeer P., Ouborg J. N., Verhoeven K. J. epiGBS: reference-free reduced representation bisulfite sequencing. (англ.) // Nature Methods. — 2016. — April (vol. 13, no. 4). — P. 322—324. — doi:10.1038/nmeth.3763. — PMID 26855363.
- Paun O., Verhoeven K. J. F., Richards C. L. Opportunities and limitations of reduced representation bisulfite sequencing in plant ecological epigenomics. (англ.) // The New Phytologist. — 2019. — January (vol. 221, no. 2). — P. 738—742. — doi:10.1111/nph.15388. — PMID 30121954.
- Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y. C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K. H., Feizi S., Karlic R., Kim A. R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L. H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M. Integrative analysis of 111 reference human epigenomes. (англ.) // Nature. — 2015. — 19 February (vol. 518, no. 7539). — P. 317—330. — doi:10.1038/nature14248. — PMID 25693563.
- Pennacchio L. A., Bickmore W., Dean A., Nobrega M. A., Bejerano G. Enhancers: five essential questions. (англ.) // Nature Reviews. Genetics. — 2013. — April (vol. 14, no. 4). — P. 288—295. — doi:10.1038/nrg3458. — PMID 23503198.
- Zhang Z., Pugh B. F. High-resolution genome-wide mapping of the primary structure of chromatin. (англ.) // Cell. — 2011. — 21 January (vol. 144, no. 2). — P. 175—186. — doi:10.1016/j.cell.2011.01.003. — PMID 21241889.
- Axel R. Cleavage of DNA in nuclei and chromatin with staphylococcal nuclease. (англ.) // Biochemistry. — 1975. — July (vol. 14, no. 13). — P. 2921—2925. — doi:10.1021/bi00684a020. — PMID 1148185.
- Deoxyribonuclease I (2013). Дата обращения: 26 ноября 2018.
- Zhou W., Sherwood B., Ji Z., Xue Y., Du F., Bai J., Ying M., Ji H. Genome-wide prediction of DNase I hypersensitivity using gene expression. (англ.) // Nature Communications. — 2017. — 19 October (vol. 8, no. 1). — P. 1038—1038. — doi:10.1038/s41467-017-01188-x. — PMID 29051481.
- An integrated encyclopedia of DNA elements in the human genome. (англ.) // Nature. — 2012. — Vol. 489, no. 7414. — P. 57—74. — doi:10.1038/nature11247. — PMID 22955616.
- He H. H., Meyer C. A., Hu S. S., Chen M. W., Zang C., Liu Y., Rao P. K., Fei T., Xu H., Long H., Liu X. S., Brown M. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. (англ.) // Nature Methods. — 2014. — January (vol. 11, no. 1). — P. 73—78. — doi:10.1038/nmeth.2762. — PMID 24317252.
- Tsompana M., Buck M. J. Chromatin accessibility: a window into the genome. (англ.) // Epigenetics & Chromatin. — 2014. — Vol. 7, no. 1. — P. 33. — doi:10.1186/1756-8935-7-33. — PMID 25473421.
- Giresi P. G., Kim J., McDaniell R. M., Iyer V. R., Lieb J. D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. (англ.) // Genome research. — 2007. — Vol. 17, no. 6. — P. 877—885. — doi:10.1101/gr.5533506. — PMID 17179217.
- Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. (англ.) // Nature Methods. — 2013. — December (vol. 10, no. 12). — P. 1213—1218. — doi:10.1038/nmeth.2688. — PMID 24097267.
- Schadt E. E., Banerjee O., Fang G., Feng Z., Wong W. H., Zhang X., Kislyuk A., Clark T. A., Luong K., Keren-Paz A., Chess A., Kumar V., Chen-Plotkin A., Sondheimer N., Korlach J., Kasarskis A. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. (англ.) // Genome Research. — 2013. — January (vol. 23, no. 1). — P. 129—141. — doi:10.1101/gr.136739.111. — PMID 23093720.
- Davis B. M., Chao M. C., Waldor M. K. Entering the era of bacterial epigenomics with single molecule real time DNA sequencing. (англ.) // Current Opinion In Microbiology. — 2013. — April (vol. 16, no. 2). — P. 192—198. — doi:10.1016/j.mib.2013.01.011. — PMID 23434113.
- Lluch-Senar M., Luong K., Lloréns-Rico V., Delgado J., Fang G., Spittle K., Clark T. A., Schadt E., Turner S. W., Korlach J., Serrano L. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. (англ.) // PLoS Genetics. — 2013. — Vol. 9, no. 1. — P. e1003191—1003191. — doi:10.1371/journal.pgen.1003191. — PMID 23300489.
- Murray I. A., Clark T. A., Morgan R. D., Boitano M., Anton B. P., Luong K., Fomenkov A., Turner S. W., Korlach J., Roberts R. J. The methylomes of six bacteria. (англ.) // Nucleic acids research. — 2012. — Vol. 40, no. 22. — P. 11450—11462. — doi:10.1093/nar/gks891. — PMID 23034806.
- Fang G., Munera D., Friedman D. I., Mandlik A., Chao M. C., Banerjee O., Feng Z., Losic B., Mahajan M. C., Jabado O. J., Deikus G., Clark T. A., Luong K., Murray I. A., Davis B. M., Keren-Paz A., Chess A., Roberts R. J., Korlach J., Turner S. W., Kumar V., Waldor M. K., Schadt E. E. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. (англ.) // Nature Biotechnology. — 2012. — December (vol. 30, no. 12). — P. 1232—1239. — doi:10.1038/nbt.2432. — PMID 23138224.
- Simpson J. T., Workman R. E., Zuzarte P. C., David M., Dursi L. J., Timp W. Detecting DNA cytosine methylation using nanopore sequencing. (англ.) // Nature Methods. — 2017. — April (vol. 14, no. 4). — P. 407—410. — doi:10.1038/nmeth.4184. — PMID 28218898.
- Laszlo A. H., Derrington I. M., Brinkerhoff H., Langford K. W., Nova I. C., Samson J. M., Bartlett J. J., Pavlenok M., Gundlach J. H. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. (англ.) // Proceedings Of The National Academy Of Sciences Of The United States Of America. — 2013. — 19 November (vol. 110, no. 47). — P. 18904—18909. — doi:10.1073/pnas.1310240110. — PMID 24167255.
- Jain M., Koren S., Miga K. H., Quick J., Rand A. C., Sasani T. A., Tyson J. R., Beggs A. D., Dilthey A. T., Fiddes I. T., Malla S., Marriott H., Nieto T., O'Grady J., Olsen H. E., Pedersen B. S., Rhie A., Richardson H., Quinlan A. R., Snutch T. P., Tee L., Paten B., Phillippy A. M., Simpson J. T., Loman N. J., Loose M. Nanopore sequencing and assembly of a human genome with ultra-long reads. (англ.) // Nature Biotechnology. — 2018. — April (vol. 36, no. 4). — P. 338—345. — doi:10.1038/nbt.4060. — PMID 29431738.
- Flusberg B. A., Webster D. R., Lee J. H., Travers K. J., Olivares E. C., Clark T. A., Korlach J., Turner S. W. Direct detection of DNA methylation during single-molecule, real-time sequencing. (англ.) // Nature Methods. — 2010. — June (vol. 7, no. 6). — P. 461—465. — doi:10.1038/nmeth.1459. — PMID 20453866.
- Yang Y., Scott S. A. DNA Methylation Profiling Using Long-Read Single Molecule Real-Time Bisulfite Sequencing (SMRT-BS) (англ.). — 2017. — Vol. 1654. — P. 125—134. — (Methods in Molecular Biology). — ISBN 978-1-4939-7230-2. — doi:10.1007/978-1-4939-7231-9_8.
- Strahl B. D., Allis C. D. The language of covalent histone modifications. (англ.) // Nature. — 2000. — 6 January (vol. 403, no. 6765). — P. 41—45. — doi:10.1038/47412. — PMID 10638745.
- Dodd I. B., Micheelsen M. A., Sneppen K., Thon G. Theoretical analysis of epigenetic cell memory by nucleosome modification. (англ.) // Cell. — 2007. — 18 May (vol. 129, no. 4). — P. 813—822. — doi:10.1016/j.cell.2007.02.053. — PMID 17512413.
- Teif V. B., Rippe K. Statistical-mechanical lattice models for protein-DNA binding in chromatin. (англ.) // Journal Of Physics. Condensed Matter : An Institute Of Physics Journal. — 2010. — 20 October (vol. 22, no. 41). — P. 414105. — doi:10.1088/0953-8984/22/41/414105. — PMID 21386588.
- Stricker Stefan H., Köferle Anna, Beck Stephan. From profiles to function in epigenomics (англ.) // Nature Reviews Genetics. — 2016. — 21 November (vol. 18, no. 1). — P. 51—66. — ISSN 1471-0056. — doi:10.1038/nrg.2016.138.
- Merriam Webster: Epigenomics.
- Callinan Pauline A., Feinberg Andrew P. The emerging science of epigenomics (англ.) // Human Molecular Genetics. — 2006. — 15 April (vol. 15, no. suppl_1). — P. R95—R101. — ISSN 1460-2083. — doi:10.1093/hmg/ddl095.
Литература
- Gibson Greg, Muse Spencer V. A primer of Genome Science. — 3rd edition. — Sunderland: Sinaeur Associates, 2009. — ISBN 978-0-87893-236-8.
- Russell Peter J. iGenetics: A Molecular Approach. — 3rd edition. — San Francisco: Pearson Benjamin Cummings, 2010. — ISBN 978-0-321-56976-9.