Секвенирование спаренных концов
Секвенирование спаренных концов — один из методов секвенирования ДНК нового поколения, основанный на получении и секвенировании библиотеки спаренных концевых фрагментов (англ. paired-end tags, PET), в которой короткие 5’- и 3’- концевые участки фрагментов ДНК/кДНК соединены друг с другом.
Конструирование библиотеки спаренных концевых фрагментов
Существует два основных метода создания библиотек спаренных концевых фрагментов: путём клонирования и без клонирования[1].
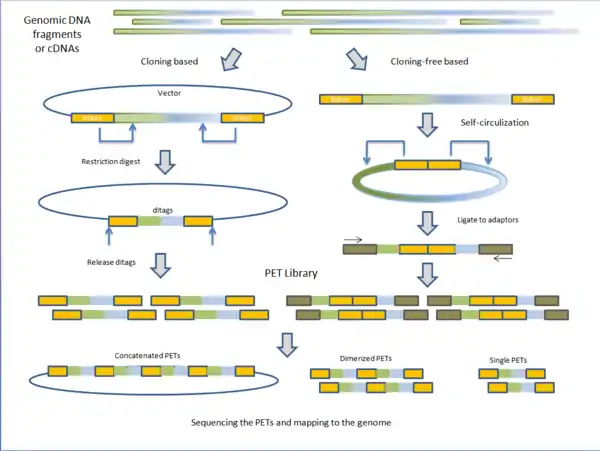
Получение путём клонирования
Геномная ДНК подвергается фрагментации (любым методом: с помощью эндонуклеаз растрикции, ультразвука, небулизацией). К фрагментам ДНК лигируют адаптеры, содержащие сайты рестрикции специальных эндонуклеаз, например, MmeI или EcoP15I. Фрагменты с адаптерами лигируют с бактериальным вектором. После этого клетки E. coli трансформируют лигазной смесью. Из полученных бактериальных колоний очищают отдельные плазмиды, обрабатывают их одной из специальных эндонуклеаз рестрикции, чьи сайты содержатся в адаптерах. Эти эндонуклеазы вырезают центральную часть клонированных фрагментов ДНК, оставляя концевые участки. После лигирования этих участков друг с другом образуются спаренные концевые фрагменты. Эти спаренные концевые фрагменты выщепляют при помощи стандартной эндонуклезаы рестрикции, сайты которой находятся по краям клонированных адаптеров. В зависимости от выбора последующей техники секвенирования, последовательности спаренных концевых фрагментов могут быть использованы в виде мономеров, димеров, или конкатемеров (несколько фрагментов, соединенные вместе).
Получение без клонирования
Фрагмент ДНК метилируют для защиты от действия эндонуклеаз рестрикции. Концы фрагмента «тупят» и фосфорилируют 5’-конец. Эти манипуляции необходимы для того, чтобы пришить адаптеры (неметилированные) на концы ДНК фрагмента. Эти адаптеры содержат сайт рестрикции, а также могут быть биотинилированы. Полученные фрагменты ДНК, фланкированные адаптерами циркуляризуются. Если адаптеры не были биотинилированы, при циклизации можно добавить биотинилированный «внутренний» адаптер. Биотин используют для изолирования целевых спаренных концевых фрагментов на сорбенте со стрептавидином. Кольцевая молекула ДНК обрабатывается эндонуклеазой — MmeI или EcoP15I, сайты связывания которых содержатся в адаптерах. Образуются свободные PET. Перед секвенированием, к этим спаренным концевым фрагментам пришивают адаптеры, которые содержат последовательности для отжига праймеров ПЦР. Полимеразная цепная реакция (ПЦР) используется для амплификации PET[2].
Преимущество создания библиотеки путём клонирования является сохранение исходных полноразмерных фрагментов кДНК. Однако клонирование длительный и трудоемкий процесс. Наибольшую популярность приобрел метод без использования клонирования. Длина последовательностей тегов спаренных концевых фрагментов может быть различна. Более длинные теги облегчают задачу картирования ридов. Эндонуклеазы, используемые для создания фрагментов описанные выше (MmeI или EcoP15I) дают теги длиной 18/20 п.н. и 25/27 п.н., соответственно[3]. Особенность этих эндонуклеаз заключаются в том, что они вносят разрыв в цепь ДНК ниже своего сайта связывания. Полученные спаренные концевые фрагменты используют для секвенирования методами следующего поколения (SOLiD, Illumina, 454 Life Sciences). Более длинные теги можно получить другими способами линеаризации ДНК после стадии циклизации фрагмента ДНК. Основными преимуществами секвенирования спаренных концевых фрагментов по сравнению с подходами, использующими один тег (т. е. тег только одного конца фрагмента ДНК) являются пониженная стоимость, повышенная специфичность картирования прочтений и возможность определения структурных особенностей генома.
Применение
Секвенирование спаренных концов ДНК
Использование спаренных концевых фрагментов для секвенирования геномов de novo имеет ряд преимуществ. Такой тип секвенирования назван попарное секвенирование концов (pairwise end sequencing), или — «double-barrel shotgun sequencing». Наибольшую популярность приобрел подход, предложенный в 1995 году[4], который являлся усовершенствованием стратегии секвенирования, описанной в 1991[5].
Технологии секвенирования следующего поколения (англ. Next-generation sequencing) позволяют прочитывать ДНК-образец весьма быстро и экономично, но длина полученных ридов гораздо меньше, по сравнению с теми, которые получают при секвенировании по методу Сенгера. Сборка геномов, в частности таких сложных, как геномы эукариот, из коротких фрагментов является комплексной проблемой. При наличии большого числа коротких последовательностей, встает вопрос о том, как их ориентировать в нужном направлении и соединить для получения полного генома. Наличие повторов в геноме еще больше осложняет эту задачу. Решением такой проблемы может служить использование спаренных концевых фрагментов.
Варьируя длину фрагмента ДНК, а значит и дистанцию между тегами можно подобрать такое расстояние, которое было бы больше повторяющего участка. В результате, картирование ридов становится однозначным. Технология секвенирования спаренных концов позволяет использовать «двусмысленные» риды (то есть те, которые картируются больше, чем на одно место в геноме) для сборки генома. Это увеличивает эффективность, снижая стоимость секвенирования, так как эти двусмысленные последовательности или риды обычно забраковываются и не рассматриваются при сборке.
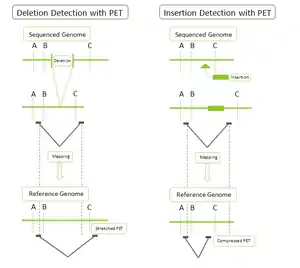
Метод секвенирования спаренных концов ДНК позволяет детектировать структурные вариации, произошедшие в геноме: вставки, делеции, инверсии и транспозиции. При создании библиотеки спаренных концевых фрагментов отбираются фрагменты ДНК равной длины, например 3 т.п.н.[6]. После выполнения оставшихся стандартных шагов (см. выше) получаем библиотеку. Секвенируем и картируем полученные риды. При картировании на референсный геном теги, полученные от одного фрагмента ДНК, должны накладываться на референсный геном на расстоянии около 3 т.п.н. (это расстояние задается при конструировании библиотеки) друг от друга и в определенной ориентации. Так, если расстояние между тегами меньше 3 т.п.н., это свидетельствует о наличие делеции в секвенируемом геноме, если больше — то о вставке. Более сложные примеры структурных вариаций генома могут быть получены при рассмотрении «противоречивых» мест картирования тегов (например, вставка последовательности из другого локуса)[2][6].
Сравнение структурных вариаций генома у двух людей (представителя африканской расы и европеоидной), показало наличие около 50 % общих структурных вариаций. «Горячие точки» структурных вариаций часто располагаются в местах генома, связанных с некоторыми заболеваниями. Структурные вариации оказывают влияние на организацию генома, так они обеспечивают перемещение экзонов, «слияние» генов, изменение ориентации гена или происходит его амплификация[6].
Метод секвенирования спаренных концов ДНК был применен также для картирования геномных перестроек раковых клеток[7].
Секвенирование спаренных концов РНК
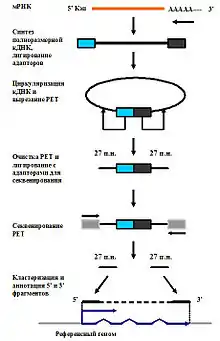
Метод используют для идентификации полноразмерных мРНК путём секвенирования 5' и 3’концевых участков соответствующей библиотеки кДНК[8][9]. На рис. 3. представлена общая схема метода. Получение библиотеки спаренных концевых фрагментов при помощи ПЦР без клонирования кДНК, позволяет включить в анализ трудно клонируемые мРНК или мРНК с очень низкой концентрацией. Далее библиотека секвенируется при помощи современных секвенаторов типа Illumina GA или SOLiD v4.
Секвенирование спаренных концов РНК используют для качественного и количественного анализа транскриптома: определения альтернативных стартов инициации транскрипции, сайтов полиаденилирования, а также определения профиля экспрессии генов. Метод также может быть использован для выявления химерных генов и случаев транс-сплайсинга, однако эти данные требуют дополнительной экспериментальной проверки.
Преимущество секвенирования спаренных концов РНК по сравнению с другими методами идентификации 5'- и 3'-концов мРНК, такими как CAGE, SAGE и SuperSAGE состоит в определении обоих концов мРНК одновременно, что обеспечивает повышенную точность картирования соответствующей мРНК на геноме. В отличие от метода полногеномного секвенирования РНК, в котором анализируется библиотека фрагментов РНК, полученных случайным образом, в секвенировании спаренных концов РНК определяют последовательности только концов молекул РНК, что существенно снижает стоимость количественного анализа транскриптома, но не дает информацию о внутренней структуре мРНК, например, о положении полиморфизмов или экзон-интронной структуре. Кроме того, стабильные вторичные структуры мРНК могут осложнить получение полноразмерной кДНК, а, значит, и идентификацию мРНК.
ChIA-PET
Анализ пространственной организации хроматина путём секвенирования спаренных концов (пер. с англ. Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET)) — молекулярно-биологический метод, позволяющий определять взаимодействия (пространственную близость) участков хроматина, расположенных на значительном удалении друг от друга в геноме. Этот метод и позволяет определить de novo пространственное расположение участков хроматина друг относительно друга. Такие взаимодействия представляют интерес для определения регуляторных элементов (например, цис-регуляторные элементы, транс-регуляторные элементы, инсуляторы, энхансеры, сайленсеры). В свою очередь, полученная информация важна для понимания механизмов регуляции экспрессии генов.
Литература
- Fullwood MJ., Wei CL, Liu ET, Ruan Y, Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses, Genome Reseach, 2009, 19:521-532
- Fei Yao, Pramila N et al., Long span DNA Paired-End-Tag (DNA-PET) sequencing strategy for the interrogation of genomic structural mutations and fusion-point-guided reconstruction of amplicons, PLos One, 2012, 7(9): e46152
- Morgan RD et al., MmeI: a minimal Type II restriction-modification system that only modifies one DNA strand for host protection, Nucleic Acids, 2008, 36(20):6558-6570
- Roach JC, Boysen C, Wang K, Hood L, Pairwise End Sequencing: A unified approach to genomic mapping and sequencing, Genomics, 1995, 26(2):345-53
- Edwards A., Caskey T., Closure strategies for random DNA sequencing, A Companion to Methods in Enzymology, 1991, pp. 41—47
- Korbel JO et al., Paired-End Mapping reveals extensive structural variation in the Human genome, Science, 2007, 318(5849): 420-6
- Campbell PJ et al., Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing, Nat Genet, 2008, 40(6): 722—729
- Ng P, Wei CL, Sung WK et al., Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation, Nat. Methods, 2005, 2: 105—111
- Ruan X, Ruan Y, Genome wide full-length transcript analysis using 5’ and 3’ paired-end-tag next generation sequencing (RNA-PET), Methods Mol.Biol., 2012, 809: 535—562