Методы секвенирования нового поколения
Секвенирование нового поколения (англ. next generation sequencing, NGS) — группа методов определения нуклеотидной последовательности ДНК и РНК для получения формального описания её первичной структуры. Технология методов секвенирования нового поколения позволяет «прочитать» единовременно сразу несколько участков генома, что является главным отличием от более ранних методов секвенирования. NGS осуществляется с помощью повторяющихся циклов удлинения цепи, индуцированного полимеразой, или многократного лигирования олигонуклеотидов. В ходе NGS могут генерироваться до сотен мегабаз и гигабаз нуклеотидных последовательностей за один рабочий цикл[1].
История секвенирования

Первая концепция секвенирования была предложена Сенгером в 1977 году[2]. Технология получила название «метод обрыва цепи». В том же году Максам и Гилберт предложили альтернативный метод, получивший название «метод химической деградации» — в его основе лежит расщепление меченого по одному концу фрагмента ДНК под действием специфических реагентов. Определение нуклеотидной последовательности проводится методом электрофореза в полиакриламидном геле с последующей авторадиографией. Необходимость в массовом, качественном и быстром секвенировании стимулировала многочисленные модификации и всевозможные улучшения этих методов. В той или иной степени изменениям подверглись практически все составляющие этого процесса. Переломной точкой развития технологии стало появление ПЦР (середина 1980-х) и автоматизация основных этапов «чтения» ДНК, давшие начало методам секвенирования следующего поколения. Платформы для методов нового поколения основываются на распараллеливании процесса «чтения» ДНК, и таким образом за один прогон работы секвенатора можно определить первичные структуры нескольких участков генома. Секвенаторы нового поколения стали значительно дешевле и гораздо эффективнее своих предшественников. На сегодняшний день производительность некоторых секвенаторов измеряется уже сотнями миллиардов пар оснований, что, например, позволяет подобным приборам сканировать индивидуальный геном человека всего за несколько дней[3].
Методы
Ниже приведены методы NGS в хронологическом порядке. Первые методы, например на основе пиросеквенирования, дали начало развитию NGS, однако практически не используются на данный момент. Остальные ниже рассмотренные методы широко используются на данный момент, каждый метод обладает своими преимуществами и спецификой применения[4][5][6].
| метод | принцип | максимальная длина прочтения, пар оснований | стоимость секвенирования 1 млн пар оснований | стоимость секвенатора | время работы за цикл | количество прочтений за цикл | преимущества | недостатки |
|---|---|---|---|---|---|---|---|---|
| 454 Life Sciences | пиросеквенирование и люцифераза | 1000 | $10 | $500 000 | 7 часов | 1 000 000 | длина прочтённых геномных участков; скорость | стоимость; погрешность |
| Illumina-SOLEXA | нуклеотиды с флуорофором и снимаемыми терминаторами | 300 | $0,05—0,15 | $1 000 000 -(NovaSeq 6000)
$100 000 -(MiSeq) |
4 часа — 55 часов | до 5 000 000 000 | эффективность, стоимость | скорость |
| SOLiD | лигирование олигонуклеотидных зондов с флуорофором | 75 | $0,13 | $595 000 | до 10 дней | до 2 400 000 000 | стоимость | скорость |
| Helicos | нуклеотиды с флуорофором и снимаемыми терминаторами | 2900 | $2 | $1 350 000 | 1 час | 35 000—75 000 | длина прочтённых геномных участков; скорость | низкая производительность при желаемой малой погрешности; стоимость |
| IonTorrent | изменение pH в процессе присоединения нуклеотидов | 600 | $1 | $100000 | 3 часа | до 5 000 000 | стоимость; скорость | погрешность |
| PacBio Sequel[9] | нуклеотиды с флуорофором | 20 000 | $2 | $600 000 | 20—30 часов | До 500 000 | длина прочтений, точность | количество материала, цена |
| MinION Mk1B[10][11] | изменение силы тока по мере прохождения цепи через нанопору | длина всей НК, до 2 000 000 | $0,47—0,90 | $1000 | 1 мин — 2 суток | — | длина прочтений, стоимость, отсутствие амплификации и сложных химических превращений | погрешность |
В связи со стремительным развитием методов секвенирования, параметры методов, такие как стоимость секвенаторов и их работы, время и длины прочтённых участков могут меняться[5].
Массивно-параллельное опознавательное секвенирование (MPSS)
Массивно-параллельное опознавательное секвенирование (англ. massively parallel signature sequencing, MPSS) — одна из первых технологий NGS, которая была разработана в 1990-х компанией Lynx Therapeutics для секвенирования мРНК транскриптов и оценки генной экспрессии, основываясь на индивидуальных уровнях мРНК в отдельной клетке[12]. В методе MPSS транскрипты захватываются на отдельных микрогранулах с матрицей ДНК; мРНК прочитываются гибридизацией с флуоресцентной меткой, а затем удаляются, и так несколько раз подряд. В результате получаются последовательности длиной от 17 до 20 пар оснований (п. о.). Количество транскриптов, показывающих уровень экспрессии, определяется числом транскриптов на миллион молекул. Данный метод не требует идентификации генов перед началом анализа, а его чувствительность — несколько молекул мРНК на клетку[13].
Roche/454 Life Sciences
Первая эффективно используемая на коммерческой основе платформа NGS. Компания 454 Life Sciences основана в 2000 году Джонатаном Ротбергом (в производство запущена в 2005 году). Данная технология представляет собой последовательный синтез методов эмульсионного ПЦР и пиросеквенирования[14].
Амплификация ДНК проходит в каплях воды в масляной эмульсии. В каждой капле воды находится одноцепочечная матрица ДНК, связанная с праймером на бусинке. Далее, каждая бусина помещается на чип, представляющий собой оптическое волокно. Туда же помещаются необходимые для секвенирования ферменты: ДНК-полимераза, люцифераза, АТФ-сульфурилаза. В последней сборке реакция секвенирования идёт в ячейках объёмом 3,4·106 пл, на стенках которых есть специальное металлическое покрытие, нивелирующее шум[15].
Illumina/Solexa
Авторы метода — британские химики Шанкар Баласубраманиан и Дэвид Кленерман. Этот метод секвенирования использует прикреплённые к микросферам единичные молекулы ДНК. В 2006 году была запущена Solexa Genome Analyzer 1G, первая платформа, генерирующая короткие участки генома. После её приобретения компанией Illumina Genome Analyzer использует оптически прозрачные ячейки с 8 индивидуальными поверхностями (иногда меньше: 4, 2 или даже 1), где связываются олигонуклеотиды. В отличие от пиросеквенирования удлинение последовательности происходит постепенно, что позволяет за раз с помощью камеры снимать большие ДНК-чипы[16].
Applied Biosystems/SOLiD
Платформа SOLiD (Supported Oligonucleotide Ligation and Detection System 2.0), разработанная Applied Biosystems — технология секвенирования коротких чтений, основанная на лигировании. Метод был предложен в лаборатории Джорджа Черча и опубликован в 2005 году. Суть метода заключается в определении нуклеотидной последовательности небольших фрагментов (25—75 п. о.) геномной ДНК; к обоим концам предварительно фрагментированной ДНК лигируют адаптеры, необходимые для эмульсионной ПЦР на магнитных шариках и последующего секвенирования на проточной ячейке[17].
Полони-секвенирование
Технология NGS без электрофоретического разделения, позволяющая прочитывать миллионы коротких иммобилизованных последовательностей ДНК. Основная идея метода — генерация большого числа уникальных «полоний» (молекулярных колоний, сгенерированных полимеразой), которые секвенируются в случайном порядке. Полони-секвенирование проводится для библиотеки парных концевых тэгов (paired-end tags): каждая молекула ДНК имеет длину 135 пар оснований (п. о.), содержит два тэга длиной 17-18 п. о., разделённых и фланкированных общей последовательностью[18][19].
Одномолекулярное секвенирование
Первый метод секвенирования единичных молекул, разработанный HeliScope (Helicos BioSciences), имеет производительность около 1 Гб/день. Принцип работы: после клональной амплификации образца происходит фрагментация ДНК с последующим полиаденилированием на 3'-конце с дальнейшим секвенированием, чередующимся с промыванием образцов нуклеотидами с флуоресцентной меткой[20]. В 2012 году компания была признана банкротом и пректратила существование[21], однако компания SeqLL, основанная в 2013 году, получила лицензию на технологию[22].
DNA nanoball sequencing

В данном методе в секвенируемый фрагмент ДНК последовательно вносятся 4 адаптера, благодаря которым в ходе дальнейшей репликации Phi29 ДНК-полимеразой (rolling circle replication) синтезируемая молекула ДНК сворачивается в ДНК-наношарики. Затем наношарики наносятся на субстрат, имеющий многочисленные поля размером ~300 нм для связывания ДНК, организованные в виде решётки. Организация этих полей позволяет уместить больше ДНК на субстрат и увеличить плотность информации в изображении по сравнению со случайным нанесением ДНК на субстрат (например, как в полони-секвенировании)[23].
cPAL
Combinatorial probe anchor ligation — комбинированный метод секвенирования, использующий комбинацию гибридизации и лигирования пула зондов. Каждый зонд состоит из девяти оснований, которые вырождены (то есть могут быть любыми из четырёх) во всех, кроме одной позиции, которую собираются прочитать. Интересующая позиция помечена одним из четырёх красителей, соответствующие каждому азотистому основанию. На матрицу гибридизируют якорную последовательность, комплементарную адаптеру и зонды. Зонды, гибридизованные напротив одного из концов якорной последовательности, затем лигируются. После гибридизации и лигирования избыток зондов смывают и снимают изображение. Затем смывают весь якорно-зондовый комплекс и процесс повторяют, используя зонды для других позиций. После считывания 5 смежных оснований процесс повторяется с использованием якорей с пятью дополнительными вырожденными основаниями, что позволяет секвенировать до 10 оснований с каждой стороны адаптера. В общей сложности секвенируется прочтение в 70 оснований из исходного фрагмента, по 35 оснований на каждом конце адаптера. Из-за расстояния между адаптерами эти последовательности в 35 оснований не являются смежными, поскольку они содержат гэп в два основания и гэп в пять оснований[24].
Ion Torrent Sequencing
Метод основан на связи между химической и цифровой информацией; эта технология также называется рН-индуцированным секвенированием. Процесс основан на детекции протонов, которые получаются при синтезе цепи ДНК как побочный продукт. Как следствие, рН раствора меняется, что и можно детектировать[25].
Платформа Ion Torrent отличается от остальных технологий секвенирования тем, что в ней не используются модифицированные нуклеотиды и оптические методы. Метод Ion Torrent позволяет исследовать транскриптомы, малые РНК, проводить ChIP-seq. Более того, с его помощью можно изучать геномы микробных сообществ[25].
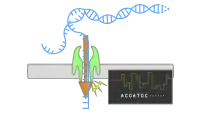
Одномолекулярное секвенирование в реальном времени Pacific Biosciences
Появление метода одномолекулярного секвенирования в реальном времени (англ. Single molecule real time sequencing, SMRT) дало возможность наблюдать за работой ДНК-полимеразы, наращивающей синтезируемую цепь, в реальном времени. Суть метода заключается в определении нуклеотидной последовательности фрагментов геномной ДНК с лигированными к их концам специфическими ДНК-адаптерами, необходимыми для последующего секвенирования. Смысл SMRT-секвенирования схож с описанными ранее методами NGS — ДНК-полимераза достраивает вторую цепь исследуемой молекулы ДНК, используя нуклеотиды, меченные различными флуоресцентными метками, которые регистрируют при помощи конфокальной микроскопии высокого разрешения[26].
Нанопоровое секвенирование
Метод основан на измерении тока ионов через единичную нанопору в непроводящей мембране. При прохождении через эту пору нуклеотидов ток падает. Время, на которое изменяется ток ионов, и величина этого падения зависят от того, какой нуклеотид в данный момент находится внутри поры[27].
Применение секвенирования нового поколения
Быстрота и дешевизна методов NGS, недоступная ранее, спровоцировала бум в индустрии геномных исследований. Благодаря NGS появилась возможность делать ранее технически недоступные эксперименты[28][29]. Применение NGS не ограничивается определением геномных последовательностей, а распространяется до изучения траскпритома, структуры хроматина и других областей молекулярной и клеточной биологии. Ниже представлены основные примеры направлений применения методов NGS[30].
ChiP-seq
Удешевление и распространение NGS позволило определять места связывания белков с ДНК (ChIP-seq), взаимодействующие участки ДНК (определение конформации хромосом) и участки открытого хроматина на протяжении всего генома, а также осуществлять проекты ENCODE и modENCODE[31].
ChiP-seq используется для картирования сайтов связывания ДНК-связывающих белков, что раньше достигалось методами иммунопреципитации хроматина и гибридизации без секвенирования на микрочипах[32].
Геномный анализ
Стали доступны геномы разных по сложности живых систем от микроорганизмов до человека, включая геном цитогенетически находящихся в норме клеток миелоидной лейкемии. Увеличение длины чтений ускорило сборку целых геномов[33].
Направленное пересеквенирование геномов
Секвенирование определённых регионов в геномах используется для выявления полиморфизмов (в частности однонуклеотидных полиморфизмов) и мутаций в генах, задействованных в развитии опухолевых и других заболеваний. Примером одной из таких широкомасштабных работ может служить проект «1000 геномов»[34].
Метагеномика
NGS широко используется в исследованиях разнообразия микроорганизмов в различных образцах (например, микробные популяции в океане и почве, идентификация новых вирусов в органах, подлежащих трансплантации, описание характерной для ЖКТ микрофлоры и т. д.)[35].
Секвенирование транскриптома
На основе NGS создан новый подход РНК-секвенирования (RNA-seq) для картирования и подсчёта транскриптов в биологических образцах. У этого метода есть преимущества над используемым ранее методом ДНК-микрочипов. Например, ДНК-чипы зависят от перекрытия геномных последовательностей, в то время как RNA-seq позволяет охарактеризовать транскрипцию без предварительного знания места начала транскрипции[36].
Перспективы применения секвенирования в медицине
В недалёком будущем технологии секвенирования станут более быстрыми и менее дорогими, что позволит использовать их для идентификации мишеней для лекарственной терапии онкологических больных. Уже в 2013 году анализ по технологии секвенирования следующего поколения от момента биопсии до завершения NGS занимал менее 100 дней. Столько же времени занимает секвенирование всего генома (whole genome sequencing, WGS) и секвенирование всего транскриптома (whole transcriptome sequencing, WTS)[37].
Примечания
- Voelkerding K. V., Dames S. A., Durtschi J. D. Next-generation Sequencing: From Basic Research to Diagnostics (англ.) // Clinical Chemistry. — 2009. — 26 February (vol. 55, no. 4). — P. 651—658. — doi:10.1373/clinchem.2008.112789.
- Ansorge W. J. Next-generation DNA sequencing techniques. (англ.) // New Biotechnology. — 2009. — April (vol. 25, no. 4). — P. 195—203. — doi:10.1016/j.nbt.2008.12.009.
- Kchouk M., Gibrat J. F., Elloumi M. Generations of Sequencing Technologies: From First to Next Generation (англ.) // Biology and Medicine. — 2017. — 6 March (vol. 9, no. 3). — doi:10.4172/0974-8369.1000395. Архивировано 26 апреля 2019 года.
- Goodwin, S., McPherson, J., McCombie, W. Coming of age: ten years of next-generation sequencing technologies. (англ.) // Nat Rev Genet. — 2016. — 17 May (no. 17). — P. 333—351. — doi:10.1038/nrg.2016.49.
- Liu L., Li Y., Li S., Hu N., He Y., Pong R., Lin D., Lu L., Law M. Comparison of Next-Generation Sequencing Systems (англ.) // Journal of Biomedicine and Biotechnology. — 2012. — 5 July (vol. 2012). — doi:10.1155/2012/251364. Архивировано 21 апреля 2020 года.
- Ребриков Д. В., Коростин Д. О., Шубина Е. С., Ильинский В. В. NGS. Высокопроизводительное секвенирование. — Бином, 2014. — 232 с. — ISBN 978-5-9963-1784-4.
- Quail M. A., Smith M., Coupland P. et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers (англ.) // BMC Genomics. — 2012. — 24 July (vol. 13, no. 341). — doi:10.1186/1471-2164-13-341. Архивировано 30 марта 2019 года.
- Barba M., Czosnek H., Hadidi A. Historical Perspective, Development and Applications of Next-Generation Sequencing in Plant Virology (англ.) // Viruses. — 2014. — 6 January (vol. 6). — P. 106—136. — doi:10.3390/v6010106. Архивировано 2 июня 2018 года.
- PacBio Sequel Systems. www.pacb.com. Дата обращения: 19 апреля 2020. Архивировано 27 апреля 2020 года.
- Product comparison (англ.). Oxford Nanopore Technologies. Дата обращения: 19 апреля 2020. Архивировано 3 декабря 2019 года.
- Branton D., Deamer D. W., Marziali A., Bayley H., Benner S. A. The potential and challenges of nanopore sequencing (англ.) // Nature biotechnology. — 2008-10. — Vol. 26, iss. 10. — P. 1146–1153. — ISSN 1087-0156. — doi:10.1038/nbt.1495. Архивировано 18 мая 2019 года.
- Schuler group. Massively Parallel Signature Sequencing (MPSS) (англ.).
- Brenner S., Johnson M., Bridgham J., Golda G., Lloyd D. H. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays (англ.) // Nature Biotechnology. — 2000-06. — Vol. 18, iss. 6. — P. 630–634. — ISSN 1087-0156. — doi:10.1038/76469. Архивировано 27 октября 2016 года.
- Zheng, Z; et al. Titration-free massively parallel pyrosequencing using trace amounts of starting material. (англ.) // Nucleic Acids Res.. — 2010. — Vol. 38, no. 13. — P. e137. — doi:10.1093/nar/gkq332. — PMID 20435675.
- Ronaghi M., Karamohamed S., Pettersson B., Uhlén M., Nyrén P. Real-Time DNA Sequencing Using Detection of Pyrophosphate Release (англ.) // Analytical Biochemistry. — 1996-11. — Vol. 242, iss. 1. — P. 84–89. — doi:10.1006/abio.1996.0432.
- An Introduction to Next-Generation Sequencing Technology (англ.). Illumina. Дата обращения: 30 апреля 2013. Архивировано 11 апреля 2013 года.
- SOLiD System (англ.). Applied Biotechnologies. Дата обращения: 5 мая 2020. Архивировано 26 марта 2020 года.
- Mitra R. D., Shendure J., Olejnik J., Edyta-Krzymanska-Olejnik, Church J. M. Fluorescent in situ sequencing on polymerase colonies (англ.) // Analytical Biochemistry. — 2003-09-01. — Vol. 320, iss. 1. — P. 55–65. — ISSN 0003-2697. — doi:10.1016/s0003-2697(03)00291-4. Архивировано 5 сентября 2015 года.
- Shendure J., Porreca G. J., Reppas N. B., Lin X., McCutcheon J. P. Accurate multiplex polony sequencing of an evolved bacterial genome (англ.) // Science (New York, N.Y.). — 2005-09-09. — Vol. 309, iss. 5741. — P. 1728–1732. — ISSN 1095-9203. — doi:10.1126/science.1117389. Архивировано 17 июля 2016 года.
- Pareek C. S., Smoczynski R., Tretyn A. Sequencing technologies and genome sequencing. (англ.) // J Appl Genetics. — 2011. — 23 June (no. 52). — P. 413—435. — doi:10.1007/s13353-011-0057-x. Архивировано 13 июня 2018 года.
- Battered Helicos BioSciences Corporation Files for Chapter 11 (англ.). BioSpace. Дата обращения: 24 мая 2020.
- About us – SeqLL (англ.). Дата обращения: 24 мая 2020.
- Drmanac R., Sparks A. B., Callow M. J., Halpern A. L., Burns N. L. Human Genome Sequencing Using Unchained Base Reads on Self-Assembling DNA Nanoarrays (англ.) // Science. — 2010-01-01. — Vol. 327, iss. 5961. — P. 78–81. — ISSN 1095-9203 0036-8075, 1095-9203. — doi:10.1126/science.1181498.
- Complete Genomics (англ.). AllSeq.
- Rusk Nicole. Torrents of sequence (англ.) // Nature Methods. — 2010. — 20 December (vol. 8, no. 1). — P. 44—44. — ISSN 1548-7091. — doi:10.1038/nmeth.f.330.
- Eid J., Fehr A., Gray J., Luong K., Lyle J., et al. Real-Time DNA Sequencing from Single Polymerase Molecules (англ.) // Science. — 2009. — 2 January (vol. 323, no. 5910). — P. 133—138. — doi:10.1126/science.1162986. Архивировано 8 сентября 2017 года.
- Eisenstein M. Oxford Nanopore announcement sets sequencing sector abuzz. (англ.) // Nat Biotechnol. — 2012. — Vol. 30. — P. 295—296. — doi:10.1038/nbt0412-295.
- Shendure J., Ji H. Next-generation DNA sequencing. (англ.) // Nature Biotechnologies. — 2008. — 9 October (no. 26). — P. 1135—1145. — doi:10.1038/nbt1486. Архивировано 11 декабря 2019 года.
- Pavlopoulos G. A. , Oulas A., Lacucci E., et al. Unraveling genomic variation from next generation sequencing data. (англ.) // BioData Mining. — 2013. — No. 6:13. — doi:10.1186/1756-0381-6-13.
- Applications of next-generation sequencing (англ.). Nature Reviews Genetics.
- S. G. Landt, G. K. Marinov, A. Kundaje, P. Kheradpour, F. Pauli. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia (англ.) // Genome Research. — 2012-09-01. — Vol. 22, iss. 9. — P. 1813–1831. — ISSN 1088-9051. — doi:10.1101/gr.136184.111.
- Bailey T., Krajewski P., Ladunga I., Lefebvre C., Li Q. Practical guidelines for the comprehensive analysis of ChIP-seq data (англ.) // PLoS computational biology. — 2013. — Vol. 9, iss. 11. — P. e1003326. — ISSN 1553-7358. — doi:10.1371/journal.pcbi.1003326. Архивировано 4 мая 2017 года.
- Henson J, Tischler G, Ning Z. 2012;13(8):901‐915. doi:10.2217/pgs.12.72. Next-generation sequencing and large genome assemblies. (англ.) // Pharmacogenomics. — 2012. — 20 March (vol. 13, no. 8). — P. 901—915. — doi:10.2217/pgs.12.72.
- Mills R., Walter K., Stewart C. et al. Mapping copy number variation by population-scale genome sequencing. (англ.) // Nature. — 2011. — 2 February (vol. 470). — P. 59—65. — doi:10.1038/nature09708. Архивировано 19 октября 2019 года.
- Eisen J. A. Environmental shotgun sequencing: its potential and challenges for studying the hidden world of microbes. (англ.) // PLoS Biol.. — 2007. — Vol. 5, no. 3. — P. e82. — doi:10.1371/journal.pbio.0050082. Архивировано 11 декабря 2019 года.
- Metzker M. Sequencing technologies — the next generation. (англ.) // Nature Reviews Genetics. — 2010. — No. 11. — P. 31—46. — doi:10.1038/nrg2626. Архивировано 6 февраля 2020 года.
- Weiss G. J., Liang W. S., Demeure M. J., Kiefer J. A., Hostetter G. et al. A Pilot Study Using Next-Generation Sequencing in Advanced Cancers: Feasibility and Challenges. (англ.) // PLoS ONE. — 2013. — 30 October (vol. 8, no. 10). — doi:10.1371/journal.pone.0076438.
