ChIA-PET
Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET) — молекулярно-биологический метод, позволяющий определять взаимодействия (пространственную близость) участков хроматина, расположенных на значительном удалении друг от друга в геноме.[1] Такие взаимодействия представляют интерес для определения регуляторных элементов. Регуляторные элементы в клетках могут находиться на значительном расстоянии от промотора регулируемого гена (например, цис-регуляторные элементы, транс-регуляторные элементы, инсуляторы, энхансеры). Для понимания механизмов таких взаимодействий необходимо знать пространственное расположение участков хроматина друг относительно друга, что этот метод и позволяет определить de novo. В свою очередь, полученная информация важна для понимания механизмов регуляции экспрессии генов.[2]
ChIA-PET основан на применении ChIР, 3С (Определение конформации хромосом) и секвенировании спаренных концов(англ. Paired-end tag), для чего используется высокопроизводительное секвенирование и дальнейшая компьютерная обработка результатов.[3]
История
Метод впервые был использован в 2009 году[1] для определения удаленных сайтов связывания ER-alpha при раке молочной железы у человека. Впоследствии был использован, например, для построения интерактома (карты возможных взаимодействий), опосредованного CTCF в эмбриональных стволовых клетках мыши[4].
Возможности метода
ChIA-PET комбинирует возможности методов, основанных на ChIP и 3C[5], расширяя возможности каждого из них. Традиционный метод ChIP позволяет определить взаимодействие определенного белка с ДНК и может использоваться для поиска сайтов связывания транскрипционных факторов. При использовании ChIP-seq могут быть определены участки связывания интересующего белка de novo в целом геноме. Если белок связывает участки хроматина, находящиеся далеко друг от друга на хромосоме, но близко в пространстве, ChIP-seq может определить каждый из них, но не укажет на их взаимодействие. При этом не все последовательности, определенные методом ChIP-seq, уникально картируются в геноме и не все являются функциональными сайтами связывания[6].
В основе методов 3C и ChIA-PET лежит теория проксимального лигирования (англ. proximity ligation), гласящая, что концы участков хроматина, связанных с белковым комплексом, находящиеся рядом, будут лигироваться друг на друга с большей вероятностью, чем концы участков, находящихся в растворе или связанных с другим белковым комплексом.
Метод 3C позволяет определить пространственную структуру хроматина, но не позволяет определить взаимодействующий белок[5]. Существенной проблемой является и необходимость точного знания последовательности взаимодействующих локусов. Это нужно для подбора праймеров для количественного или полуколичественного ПЦР-анализа, используемого для их определения. (Надо заметить, что локусов — кандидатов на взаимодействия — определяемых методом 3C , может быть несколько). Метод ChIA-PET позволяет de novo определять пространственную структуру хроматина, связанного со специфическим белком. То есть, с одной стороны, не требует знания последовательности ДНК в районе взаимодействия, и, с другой полностью зависит от специфичности используемых антител.
Сравнительные характеристики
| ChIР | ChIP-seq | ChIA-PET | 3C | |
|---|---|---|---|---|
| Необходимо иметь специфические антитела | ||||
| Необходимо знать последовательность ДНК в исследуемом локусе | ||||
| Необходимо иметь референсный геном | ||||
| Метод дает информацию о | Сайтах связывания | О сайтах связывания de novo | О сайтах связывания de novo+ конформация хроматина | Конформации хроматина |
Описание метода
Экспериментальные методы
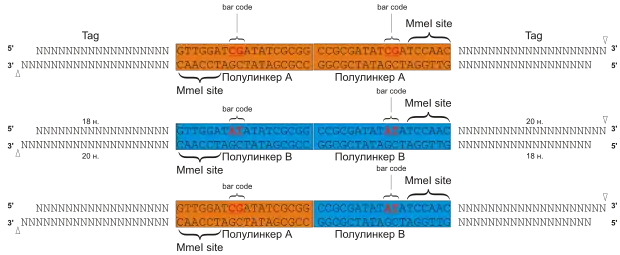
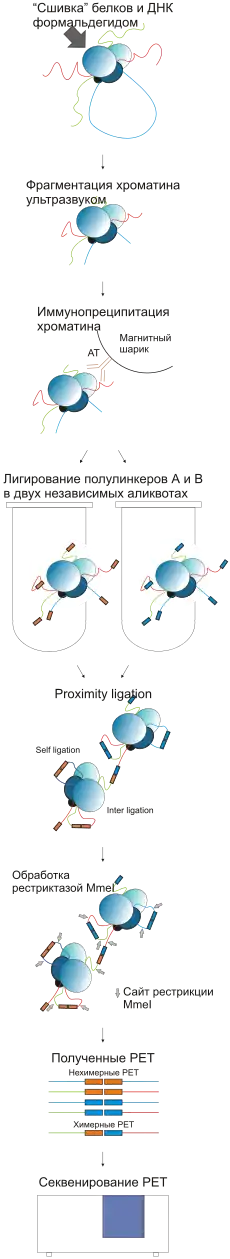
Комплексы ДНК-белок неспецифически сшиваются с помощью формальдегида. Образец подвергается воздействию ультразвука, при этом молекулы ДНК дробятся на фрагменты и разрушаются непрочно связавшиеся неспецифические комплексы. В результате получаются фрагменты ДНК в прочных комплексах с белками. Далее с помощью специфических антител, закрепленных на магнитных шариках, преципитируются фрагменты хроматина, связанные с интересующим белком. Часто объектами исследования являются известные транскрипционные факторы[1]. Преципитированные комплексы извлекаются из раствора за магнитные шарики при помощи магнита. Выделенные комплексы разделяются на 2 аликвоты и на концы молекул ДНК «пришиваются» олигонуклеотидные полулинкеры с известной последовательностью. В одной аликвоте — полулинкер А, в другой — полулинкер В. Оба полулинкера содержат сайт, узнаваемый рестриктазой MmeI, и отличаются друг от друга «баркодом» (barcode) из двух нуклеотидов: CG для полулинкера А, и AT для полулинкера В. В результате в дальнейшем при секвенировании можно отличить линкеры друг от друга по «баркоду». На следующем этапе две аликвоты объединяются и происходит проксимальное лигирование, в результате которого полулинкеры лигируются друг на друга с образованием линкеров полной длины. Линкеры с АА (CG/CG) или BB (AT/AT) «баркодами» считаются вероятными продуктами лигирования внутри одного комплекса, в то время как линкеры с АВ (CG/AT) «баркодами» считаются химерными продуктами лигирования ДНК, связанной с разными белковыми комплексами Подготовка к сиквенсу включает в себя обработку комплексов рестриктазой MmeI[8], которая расщепляет ДНК на определенном расстоянии от своего сайта узнавания в полулинкере. В результате в конце этого этапа получаются конструкции, содержащие пару «тагов» (англ. tag) (по 20 п.н. каждый) с двух сторон от полного линкера (38 п.н.). Полученные фрагменты секвенируют с обоих концов(англ. PET). После этого «таги» картируются на геном.[7][9]
Компьютерная обработка
Компьютерная обработка результатов секвенирования включает в себя 6 модулей[7][10]
Фильтрация линкеров
Все прочитанные последовательности разделяются на последовательности, имеющие читаемые «баркоды» и нечитаемые «баркоды». Если «баркод» не может быть прочитан, то последовательность «отбрасывается» из обработки. Если «баркод» может быть прочитан, то последовательности выравниваются по линкерам. Все полученные последовательности разделяются на химерные (образованные при лигировании ДНК из разных комплексов и содержащие линкер А/В) и нехимерные (содержащие линкер А/А или В/В). Надо заметить, что среди последовательностей, содержащих А/А или В/В также могут встречаться химерные. Далее последовательности собственно линкеров «отбрасываются» и анализируются последовательности «тагов» (РЕТs).[7][10]
Картирование РЕТs
Полученные на предыдущем этапе последовательности картируются в референсном геноме. На первом этапе выделяются последовательности, выравнивающиеся на 100 %, которые могут картироваться в одном локусе (уникальные) или во многих локусах. Из оставшихся последовательностей выделяют те, которые содержат 1 замену в последовательности (англ. missmatch) с референсным геномом, они также делятся на уникальные и последовательности с множественным картированием. Все остальные последовательности относятся к некартируемым. Все последовательности кроме уникально картируемых «отбразываются» из обработки.[7][10]
Классификация РЕТs
Выделяют две группы РЕТs: "самолигированные" (англ. self-ligation PETs) и "интерлигированные" (англ. inter-ligation PETs). "Самолигированные" (англ. self-ligation PETs) соответствуют концам одного СhIP-фрагмента ДНК, они должны располагаться на одной хромосоме на небольшом расстоянии друг от друга, в ориентации «голова к хвосту». "Интерлигированные" (англ. inter-ligation PETs) делятся на интрахромосомные (картируются на одной хромосоме на большом расстоянии), интерхромосомные (картируются на разных хромосомах) и лигированные в разной ориентации таги (англ. different orientation ligation PETs) (картируются на одной хромосоме на небольшом расстоянии, но в неправильной ориентации или на разных цепях ДНК). Граница, отделяющая "самолигированные" PETs от "интерлигированных" PETs, естественно определяется длиной фрагментов ДНК, получаемых при данной интенсивности «озвучивания». В разных экспериментах она составляла 3 — 4,6 Кb.[7][10]
Определение сайтов связывания белков
Для определения сайтов связывания белков используются "самолигированные" таги (англ. self-ligation PETs). Процедура проводится аналогично применяемой в ChIP-seq.
Определение хроматиновых взаимодействий
В предсказании этого вида взаимодействий используются "интерлигированные" (англ. inter-ligation PETs).
Визуализация и организация результатов
Данные из предыдущих этапов заносятся в базы данных для хранения, обработки и возможной визуализации.
Недостатки метода
- В данном методе соотношение сигнал/шум не слишком велико. Это может быть связано с неспецифичными белковыми комплексами, недостаточной специфичностью используемых антител, проведением ПЦР перед секвенированием и ошибками секвенирования. Все эти факторы могут вносить ошибки.
- Второй важный момент: необходимость отказаться от анализа большого количества данных. Например, неуникальные PETs, расположенные в повторах, где также могут присутствовать функциональные сайты связывания транскрипционных факторов[11]
- Невозможно определить, какой из белков комплекса, «сидящего» на регуляторном участке, отвечает за формирование петли ДНК. Для этого требуется проведение дополнительных экспериментов, например, 3С.
- Необходимы специфичные антитела, кроме того, белок в составе комплекса может быть экранирован и недоступен для антител, и в этом случае сайты связывания комплекса не будут определены.
- Необходимо наличие референсного генома.
Преимущества метода
- Позволяет de novo определять как хроматиновые взаимодействия, так и сайты связывания белков.
- Не требует знания последовательности ДНК в сайте связывания с белками.
- Возможность регулировать избирательность метода подбором специфических антител либо на специфичный белок (например, специфичный транскрипционный фактор) либо на повсеместно используемый белок (например, общие транскрипционные факторы).
- Совместим с tag-based NGS-секвенированием[12].
Программное обеспечение используемое для анализа результатов[13]
В ChIA-PET экспериментах используют следующие компьютерные программы
- ELAND Картирует ChIP-обогащенные фрагменты ДНК на референсный геном человека.
- Eisen software Определяет уровни экспрессии генов основываясь на иерархическом кластеринге.
- RepeatMasker In-silico маскирует повторяющиеся элементы.
- Monte Carlo simulation Используется для оценки уровня ложных результатов.[14]
- PET-Tool Программное обеспечение для процессинга и работы с Paired-End di-Tag sequence data.
- ChIA-PET Tool Программное обеспечение для процессинга ChIA-PET data.ChIA-PET Tool web site ChIA-PET Tool paper Архивная копия от 15 января 2014 на Wayback Machine
См. также
Примечания
- Melissa J. Fullwood, et al. An Oestrogen Receptor α-bound Human Chromatin Interactome. Nature. 2009 November 5; 462(7269): 58-64.
- Elzo de Wit and Wouter de Laat A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012 January 1; 26(1): 11-24.
- Melissa J. Fullwood and Yijun Ruan ChIP-based methods for the identification of long-range chromatin interactions. J Cell Biochem. 2009 May 1; 107(1): 30-39.
- Lusy Handoko et al.CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells. Nat Genet. 2011 June 19; 43(7): 630—638.
- Dekker J Capturing chromosome conformation. Science. 2002 Feb 15;295(5558):1306-11.
- Johnson et al., (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science. (316); 1497—502.
- Reviewed by Guoliang Li tn al.ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010; 11(2): R22.
- (недоступная ссылка) Описание рестриктазы на сайте www.molbiol.edu.ru
- Yufen Goh et al. Chromatin Interaction Analysis with Paired-End Tag Sequencing (ChIA-PET) for Mapping Chromatin Interactions and Understanding Transcription Regulation.J Vis Exp. 2012; (62): 3770.
- Stein L et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599-1610.
- Polak & Domany, Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics. 2006,(7); 133
- Contributed by Fullwood and Ruan Whole Genome Chromatin Interaction Analysis with Paired-End Tags (ChIA-PET) Архивная копия от 26 декабря 2010 на Wayback Machine. 2010
- en:ChIA-PET
- Monte Carlo method