Бисульфитное секвенирование
Бисульфи́тное секвени́рование — общее название группы методов, направленных на изучение паттерна метилирования ДНК посредством обработки её бисульфитом.
Метилирование — первое открытое эпигенетическое маркирование. Оно влияет на уровень экспрессии генов, подавляя транскрипционную активность. Кроме того, во многих случаях метилирование наследуемо[1][2], что придает дополнительный интерес его исследованию.
Бисульфит действует на одноцепочечную ДНК, превращая цитозин в урацил[3]. Если же этот цитозин метилирован, то есть к его пятому атому углерода присоединена метильная группа, то такой цитозин не подвергается превращению. Таким образом, бисульфит изменяет последовательность ДНК в зависимости от её паттерна метилирования, и после его воздействия можно установить, какие CpG-динуклеотиды были метилированы, сравнив измененную последовательность с исходной.
Методы
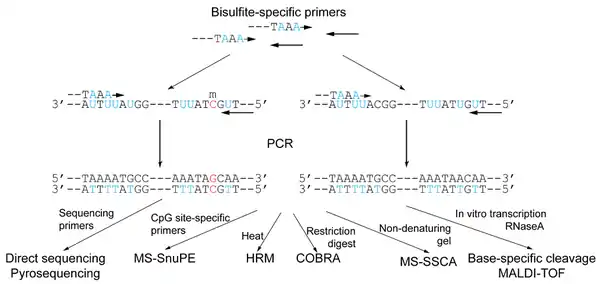
Описанные ниже методы используют секвенирование обработанных бисульфитом участков ДНК для определения паттерна метилирования. Существуют также методы, не основанные на секвенировании, например, комбинированный бисульфитный рестриктационный анализ (COBRA) и иммунопреципитация метилированной ДНК (MeDIP). Задачи изучения паттернов метилирования могут состоять как в исследовании метилирования конкретного цитозина, так и в определении доли метилированных цитозинов на каком-то участке ДНК, или даже по всему геному в целом. Из приводимых далее методов некоторые больше приспособлены для изучения конкретных сайтов метилирования, другие же — для изучения метилирования на более высоких уровнях. В идеале необходимо определять метилирование каждого аллеля. Описанию методов бисульфитного секвенирования посвящены несколько обзоров[4][5][6][7][8].
Прямое секвенирование
Первый метод бисульфитного секвенирования описан в 1992 году[9]. Для определения паттерна метилирования использовали ПЦР, праймеры для которой были специфичными как к изменённой, так и к неизменённой бисульфитом ДНК, то есть они не содержали цитозинов, входящих в CpG-динуклеотиды. Для праймеров использовали участки, близкие к интересующему сайту метилирования, но не содержащие его. В случае, когда цитозин не был метилирован, в амплифицированной последовательности обнаруживался тимин (урацил), а в синтезированной комплементарной последовательности — аденин. Если цитозин был метилирован, то в амплифицированной последовательности он и остался, а в комплементарной синтезировался гуанин. Такой метод очень трудозатратен, поскольку требует клонирования продуктов ПЦР для достижения необходимой чувствительности. Эту же проблему можно решить при помощи вложенной ПЦР.
Пиросеквенирование
В пиросеквенировании используют ПЦР с праймерами, амплифицирующими как изменённую, так и не изменённую бисульфитом ДНК. Соотношение числа метилированных и неметилированных цитозинов в амплифицируемой последовательности определяется по соотношению количества нуклеотидов А и Г в синтезируемой комплементарной последовательности[10][11].
Дальнейшее усовершенствование этого метода заключается в использовании аллель-специфических праймеров с однонуклеотидными полиморфизмами, позволяющих исследовать паттерны метилирования по отдельности в отцовских и материнских аллелях, что особенно полезно при изучении геномного импринтинга[12].
Чувствительный к метилировании анализ одноцепочечных конформаций
Этот метод основан на методе анализа одноцепочечных конформационных полиморфизмов (англ. single-strand conformation polymorphism analysis, SSCA). SSCA был разработан для выявления однонуклеотидных полиморфизмов (SNP)[13]. Фрагменты ДНК одинаковой длины, но разной нуклеотидной последовательности, с разной скоростью перемещаются в электрофорезе. Вообще говоря, SSCA недостаточно чувствителен для выявления единичной замены, но при достаточном количестве полиморфизмов неметилированных CpG-динуклеотидов в обработанной и необработанной бисульфитом ДНК будет достаточно много, чтобы чувствительности метода оказалось достаточно для определения доли метилированных цитозинов на рассматриваемом участке. Данный метод не позволяет исследовать метилированию в конкретном сайте, однако дает представление о метилировании на уровне некоторого участка или даже всего генома.
Методы высокочувствительного плавления
Метод высокочувствительного плавления (HRM) основан на ПЦР в реальном времени[14]. Температуру повышают с 55 до 95 °C, в результате чего комплементарные связи между цепочками разрушаются. Скорость плавления регистрируют при помощи специальных флуоресцентных красителей, и, зная зависимость флуоресценции от нуклеотидной последовательности цепочек, можно различитьоднонуклеотидные полиморфизмы. Метод не дает информации о том, какие именно CpG-динуклеотиды были метилированы и потому не изменились в процессе бисульфитной обработки, однако дает достаточно точную информацию об их количестве на интересующем участке.
Чувствительный к метилированию метод однонуклеотидного удлинения праймера
Метод однонуклеотидного расширения праймера исходно был разработан для анализа однонуклеотидных полиморфизмов[15]. Применительно к анализу метилирования его используют следующим образом. На обработанной бисульфитом одноцепочечной ДНК отжигают праймеры до пары оснований, непосредственно предшествующей интересующему сайту метилирования. Затем праймер удлиняетсяпри помощи ДНК-полимеразы с использованием терминирующих её дидезоксинуклеотидов. Соотношение числа метилированных и неметилированных цитозинов в исходной последовательности определяется по соотношению количеств расширений праймеров нуклеотидами Г и А соответственно. Определить соотношение числа метилированных и неметилированных цитозинов в исходной последовательности можно разными способами. Используют радиоактивные или флуоресцентные метки, а также пиросеквенирование[16].
Кроме того, это можно делать при помощи матрично-активированной лазерной десорбции/ионизации с последующим применением время-пролетного масс-анализатора (MALDI-TOF) или ион-парной обращённо-фазной высокоэффективной жидкостной хроматографии (IP-RP-HPLC)[17].
Расщепление, специфичное к основанию
Метод основан на использовании специфического расщепления РНК[18]. При добавлении к праймеру в ПЦР промотора РНК-полимеразы in vitro синтезируется РНК-транскрипт интересующего участка, после чего расщепляется рибонуклеазой А. Рибонуклеаза А расщепляет одноцепочечную РНК на местах расположения нуклеотидов Ц и У. Используя устойчивые к расщеплению нуклеозидтрифосфаты dUTP и dCTP, можно добиться специфического расщепления в местах расположения Ц или У. Фрагменты РНК затем анализируют при помощи MALDI-TOF. Данный метод даёт информацию о метилировании каждого из имеющихся CpG-динуклеотидов, а не только о доле метилированных нуклеотидов.
Метилированно-специфичная ПЦР (MSP)
Этот метод использует праймеры, специфичные или только к преобразованной бисульфитом ДНК, или только к непреобразованной[19]. Соответственно, к первым праймерам прикрепляются только участки, которые были метилированы, а ко вторым — те, которые не были, что лишает необходимости секвенировать участки после амплификации.
Амплифицированную в MSP ДНК можно далее анализировать разными другими методами. Метод MethyLight использует MSP в реальном времени, основанную на флуоресценции][20]. Mc-MSP использует анализ кривых плавления[21]. Высокочувствительный метод плавления использует и то, и другое, и потому обладает достаточной чувствительностью для определения метилирования низкого уровня[22].
Методы, основанные на микрочипах
Использование ДНК-микрочипов дает возможность расширить описанные выше методы для анализа метилированности на уровне всего генома[23]. Олигонуклеотиды ДНК-микрочипа делают специфичными к метилированию и соответствующими интересующим CpG-сайтам. Одни комплементарны измененной бисульфитом последовательности (то есть той, в которой CpG-сайт не был метилирован), другие — неизмененной. Примером такого метода служит анализ метилирования Illumina.
Ограничения методов
Методы бисульфитного секвенирования имеют ряд ограничений. У млекопитающих широко распространена модификация ДНК 5-гидроксиметилцитозин. При обработке бисульфитом 5-гидроксиметилцитозин превращается в цитозин-5-метилсульфонат, который при секвенировании опознаётся как Ц. Таким образом, бисульфитное секвенирование не может различить 5-гидроксиметилцитозин и метилцитозин, а потому не может рассматриваться как метод, чувствительный только к метилированию ДНК. В 2012 году был разработан метод бисульфитного секвенирования, позволяющий различить эти две модификации[24].
Поскольку неполное превращение азотистых оснований в ДНК под действием бисульфита, которое возможно только в одноцепочечной ДНК, даёт неправильные результаты, необходима точная подборка условий эксперимента (температура, концентрация солей), чтобы поддерживать ДНК в денатурированном состоянии[4]. Поддержанию одноцепочечной конформации ДНК может способствовать её закрепление в агарозном геле[25].
Ещё одна проблема состоит в деградации ДНК при обработке бисульфитом. Поскольку бисульфит действует только на одноцепочечную ДНК, необходимо проводить реакцию в таких условиях, в которых ДНК оставалась бы одноцепочечной, причем проводить её достаточное для успешной конвертации время. Однако, такие условия, а именно высокая температура и длительный период инкубации, могут привести к деградации до 90 % всей ДНК[26].
Примечания
- Lunerová-Bedrichová J., Bleys A., Fojtová M., Khaitová L., Depicker A., Kovarík A. Trans-generation inheritance of methylation patterns in a tobacco transgene following a post-transcriptional silencing event. (англ.) // The Plant journal : for cell and molecular biology. — 2008. — Vol. 54, no. 6. — P. 1049—1062. — doi:10.1111/j.1365-313X.2008.03475.x. — PMID 18315537.
- Ou X., Zhang Y., Xu C., Lin X., Zang Q., Zhuang T., Jiang L., von Wettstein D., Liu B. Transgenerational inheritance of modified DNA methylation patterns and enhanced tolerance induced by heavy metal stress in rice (Oryza sativa L.). (англ.) // Public Library of Science ONE. — 2012. — Vol. 7, no. 9. — P. e41143. — doi:10.1371/journal.pone.0041143. — PMID 22984395.
- Hayatsu H., Wataya Y., Kai K., Iida S. Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. (англ.) // Biochemistry. — 1970. — Vol. 9, no. 14. — P. 2858—2865. — PMID 5459538.
- Fraga M. F., Esteller M. DNA methylation: a profile of methods and applications. (англ.) // BioTechniques. — 2002. — Vol. 33, no. 3. — P. 632—634. — PMID 12238773.
- El-Maarri O. Methods: DNA methylation. (англ.) // Advances in experimental medicine and biology. — 2003. — Vol. 544. — P. 197—204. — PMID 14713229.
- Laird P. W. The power and the promise of DNA methylation markers. (англ.) // Nature reviews. Cancer. — 2003. — Vol. 3, no. 4. — P. 253—266. — doi:10.1038/nrc1045. — PMID 12671664.
- Reinders J., Paszkowski J. Bisulfite methylation profiling of large genomes. (англ.) // Epigenomics. — 2010. — Vol. 2, no. 2. — P. 209—220. — doi:10.2217/epi.10.6. — PMID 22121871.
- Wojdacz T. K., Møller T. H., Thestrup B. B., Kristensen L. S., Hansen L. L. Limitations and advantages of MS-HRM and bisulfite sequencing for single locus methylation studies. (англ.) // Expert review of molecular diagnostics. — 2010. — Vol. 10, no. 5. — P. 575—580. — doi:10.1586/erm.10.46. — PMID 20629507.
- Frommer M., McDonald L. E., Millar D. S., Collis C. M., Watt F., Grigg G. W., Molloy P. L., Paul C. L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 1992. — Vol. 89, no. 5. — P. 1827—1831. — PMID 1542678.
- Colella S., Shen L., Baggerly K. A., Issa J. P., Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. (англ.) // BioTechniques. — 2003. — Vol. 35, no. 1. — P. 146—150. — PMID 12866414.
- Tost J., Dunker J., Gut I. G. Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. (англ.) // BioTechniques. — 2003. — Vol. 35, no. 1. — P. 152—156. — PMID 12866415.
- Wong H. L., Byun H. M., Kwan J. M., Campan M., Ingles S. A., Laird P. W., Yang A. S. Rapid and quantitative method of allele-specific DNA methylation analysis. (англ.) // BioTechniques. — 2006. — Vol. 41, no. 6. — P. 734—739. — PMID 17191619.
- Bianco T., Hussey D., Dobrovic A. Methylation-sensitive, single-strand conformation analysis (MS-SSCA): A rapid method to screen for and analyze methylation. (англ.) // Human mutation. — 1999. — Vol. 14, no. 4. — P. 289—293. — doi:10.1002/(SICI)1098-1004(199910)14:4<289::AID-HUMU3>3.0.CO;2-A. — PMID 10502775.
- Wojdacz T. K., Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. (англ.) // Nucleic acids research. — 2007. — Vol. 35, no. 6. — P. e41. — doi:10.1093/nar/gkm013. — PMID 17289753.
- Gonzalgo M. L., Jones P. A. Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). (англ.) // Nucleic acids research. — 1997. — Vol. 25, no. 12. — P. 2529—2531. — PMID 9171109.
- Uhlmann K., Brinckmann A., Toliat M. R., Ritter H., Nürnberg P. Evaluation of a potential epigenetic biomarker by quantitative methyl-single nucleotide polymorphism analysis. (англ.) // Electrophoresis. — 2002. — Vol. 23, no. 24. — P. 4072—4079. — doi:10.1002/elps.200290023. — PMID 12481262.
- Matin M. M., Baumer A., Hornby D. P. An analytical method for the detection of methylation differences at specific chromosomal loci using primer extension and ion pair reverse phase HPLC. (англ.) // Human mutation. — 2002. — Vol. 20, no. 4. — P. 305—311. — doi:10.1002/humu.10118. — PMID 12325026.
- Ehrich M., Nelson M. R., Stanssens P., Zabeau M., Liloglou T., Xinarianos G., Cantor C. R., Field J. K., van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2005. — Vol. 102, no. 44. — P. 15785—15790. — doi:10.1073/pnas.0507816102. — PMID 16243968.
- Herman J. G., Graff J. R., Myöhänen S., Nelkin B. D., Baylin S. B. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 1996. — Vol. 93, no. 18. — P. 9821—9826. — PMID 8790415.
- Eads C. A., Danenberg K. D., Kawakami K., Saltz L. B., Blake C., Shibata D., Danenberg P. V., Laird P. W. MethyLight: a high-throughput assay to measure DNA methylation. (англ.) // Nucleic acids research. — 2000. — Vol. 28, no. 8. — P. 32. — PMID 10734209.
- Akey D. T., Akey J. M., Zhang K., Jin L. Assaying DNA methylation based on high-throughput melting curve approaches. (англ.) // Genomics. — 2002. — Vol. 80, no. 4. — P. 376—384. — PMID 12376091.
- Kristensen L. S., Mikeska T., Krypuy M., Dobrovic A. Sensitive Melting Analysis after Real Time- Methylation Specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection. (англ.) // Nucleic acids research. — 2008. — Vol. 36, no. 7. — P. e42. — doi:10.1093/nar/gkn113. — PMID 18344521.
- Adorján P., Distler J., Lipscher E., Model F., Müller J., Pelet C., Braun A., Florl A. R., Gütig D., Grabs G., Howe A., Kursar M., Lesche R., Leu E., Lewin A., Maier S., Müller V., Otto T., Scholz C., Schulz W. A., Seifert H. H., Schwope I., Ziebarth H., Berlin K., Piepenbrock C., Olek A. Tumour class prediction and discovery by microarray-based DNA methylation analysis. (англ.) // Nucleic acids research. — 2002. — Vol. 30, no. 5. — P. e21. — PMID 11861926.
- Yu M., Hon G. C., Szulwach K. E., Song C. X., Jin P., Ren B., He C. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. (англ.) // Nature protocols. — 2012. — Vol. 7, no. 12. — P. 2159—2170. — doi:10.1038/nprot.2012.137. — PMID 23196972.
- Olek A., Oswald J., Walter J. A modified and improved method for bisulphite based cytosine methylation analysis. (англ.) // Nucleic acids research. — 1996. — Vol. 24, no. 24. — P. 5064—5066. — PMID 9016686.
- Grunau C., Clark S. J., Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. (англ.) // Nucleic acids research. — 2001. — Vol. 29, no. 13. — P. 65. — PMID 11433041.