Получение спиртов
Спирты являются обширным и очень разнообразным классом органических соединений: они широко распространены в природе, имеют важнейшее промышленное значение и обладают исключительными химическими свойствами.
Существует огромное количество методов получения спиртов, при этом их можно разделить на две условных группы:
- химические способы получения спиртов — синтетические спирты;
- биохимические способы получения спиртов — биоспирты.
Химические способы получения спиртов
Занимая одну из центральных позиций в органической химии, спирты могут быть получены из множества других соединений. На практике в качестве исходных веществ для синтеза спиртов наиболее часто используют [1][2] :
- алкилгалогениды — щелочной гидролиз или реакция с супероксидом калия;
- алкены — кислотная гидратация, реакция гидроксимеркурирования-демеркурирования или гидроборирование с последующим окислением, а также промышленные методы оксо-синтеза;
- карбонильные соединения — восстановление или взаимодействие с реактивами Гриньяра.
Далее в разделе будет подробно рассмотрена химия существующих методов получения одноатомных спиртов. Промышленные аспекты получения спиртов, включая биохимические методы синтеза, подробно рассмотрены в подразделе «Получение спиртов в промышленности».
Краткий обзор методов органического синтеза многоатомных спиртов будет рассмотрен в соответствующем подразделе.
Получение спиртов из галогеноуглеводородов
Галогенпроизводные углеводородов под действием оснований трансформируются с образованием спиртов (реакция нуклеофильного замещения).
Обычно, первичные и вторичные галогенуглеводороды вступают в реакцию по одностадийному SN2 механизму[3]. Пример — гидролиз бромэтана:
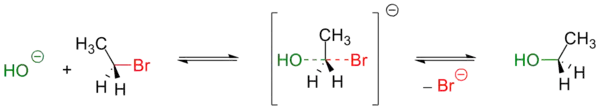
Реакции такого типа, обычно, происходят — с обращением геометрической конфигурации исходного вещества[3]. Реакционная способность алкинов уменьшается при переходе от производных йода к производным фтора [4] При этом фторпроизводные устойчивы к нуклеофильному замещению в обычных условиях и практически не используются для получения спиртов.
Первичные хлоралканы удовлетворительно гидролизуются под действием водного раствора щёлочи при нагревании[5]:

Для реакций, протекающих по SN2 механизму, используют только полярные растворители, причем скорость превращения возрастает при использовании вместо протонных растворителей (например: вода или спирт) апротонные (например: диметилсульфоксид); при этом в апротонных растворителях нуклеофильность уходящих групп будет иной[3]:

Третичные и в меньшей степени вторичные галогенуглеводороды гидролизуются по двухстадийному SN1 механизму[3]:
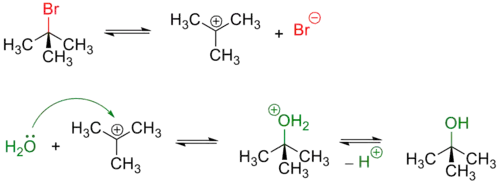
Реакция, протекающая по SN1 механизму проводят в полярных протонных растворителях, чаще всего воде или водном растворе метилового или этилового спирта.
Из-за устойчивости карбкатиона по такому механизму гидролизуются галогеналкены:

Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учёта фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта.
Для высокореакционных реагентов используют мягкое замещение с использованием соединений одновалентного серебра или двухвалентной ртути[5]:
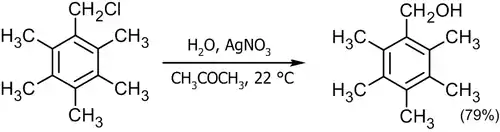

В современной лабораторной практике описанные выше реакции сольволиза проводят достаточно редко, так как спирты — более доступные полупродукты — являются исходным объектом для синтеза галогенпроизводных. Кроме того, следует помнить, что помимо изменения стереохимии исходных компонентов, реакции замещения конкурируют с элиминированием, а также перегруппировками, что часто приводит к нежелательным продуктам[3]:


В то же время существует достаточно новый метод превращения в спирты алкилгалогенидов действием на последние супероксида калия в среде диметилсульфоксида в присутствии 18-краун-6, при этом происходит практически полное геометрическое обращение[2]:
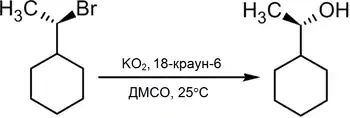 Гидролиз алкилгалогенидов с применением суперокисда калия |
Гидратация алкенов
Кислотная гидратация алкенов исторически была первым синтетическим методом получения спиртов (см. подраздел «История открытия спиртов»).
Общий механизм процесса (реакция электрофильного присоединения AdE2) представлен ниже[6]:


Присоединение происходит по правилу Марковникова.
В случае использования серной кислоты в качестве катализатора промежуточным продуктом является эфир серной кислоты (R-CH(OSO2OH)-CH3), который в условиях реакции полностью гидролизуется до спирта[6].
Для проведения реакции кроме серной кислоты используют и другие реагенты: смесь муравьиной и каталитического количества серной кислоты (в отдельных случаях позволяет добиться стереоспецифичности), смесь муравьиной и хлорной кислоты, трифторуксуную кислоту и др[7].
Реакции вторичных алкенов, вследствие перегруппировок карбокатионов, часто приводят к образованию смеси продуктов, что затрудняет их использование для получения вторичных спиртов[8]:

В лабораторной практике метод кислотной гидратации применим весьма ограниченно как из-за перспективы получения смеси продуктов, так и низких выходов. Чаще его используют для получения третичных спиртов, но и в этом случае выход, обычно, не превышает 40-45 %[8]:

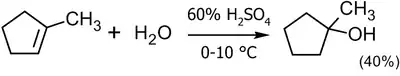 Кислотная гидратация |
В промышленности, помимо жидкофазной используют прямую газофазную гидратацию алкенов. В качестве катализаторов используется фосфорная кислота на твердом носителе при 200—300 °C и давлении 2-8 МПа; при этом выход спиртов достигает 95 %[9]:


Гидроксимеркурирование-демеркурирование алкенов
Метод получения спиртов гидроксимеркурированием-демеркурированием алкенов имеет ряд важных преимуществ перед реакцией кислотного гидролиза[10]:
- отсутствие перегруппировок для склонных для этого субстратов;
- анти-стереоспецифичность для нормальных алкенов за исключением особых случаев пространственного затруднения;
- лучшие выходы;
- строгая ориентация по правилу Марковникова.
Механизм реакции выглядит следующим образом[11]:
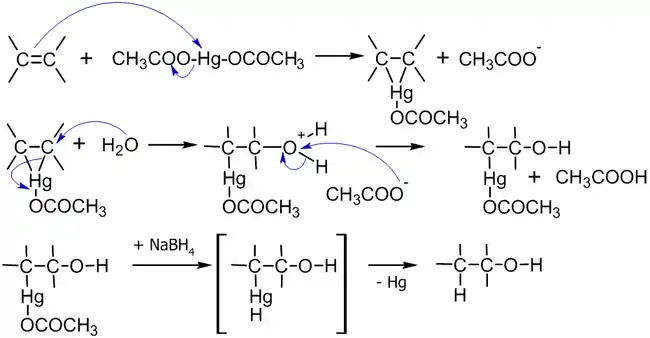 |
Присоединение ацетата ртути к алкену происходит по электрофильному механизму, а демеркурирование имеет радикальную природу; так как последняя стадия не обладает высокой стереоселективностью, то и весь процесс не стереоспецифичен в строгом смысле[12].
Синтез спиртов гидроксимеркурированием-демеркурированием алкенов протекает в мягких условиях, с выходами близкими к количественным (90-98 %) и практически без образования побочных продуктов; при этом промежуточное ртутьорганическое соединение не требует выделения — все стадии реакции протекают в один за другим[12].
Практические примеры использования реакции (в скобках указаны выходы, показывающие соотношение образующихся продуктов)[12]:



Гидроборирование алкенов с последующим окислением
Присоединение гидридов бора к алкенам и последующее их расщепление в щелочной среде, открытое Г. Брауном в 1958 году, является столь важной реакцией, что за её обнаружение и изучение в 1979 году учёный был удостоен Нобелевской премии по химии[13].
Присоединение происходит многоступенчато с образованием промежуточного циклического активированного комплекса, причем присоединение бора происходит против правила Марковникова — к наиболее гидрогенизированному атому углерода[14]:
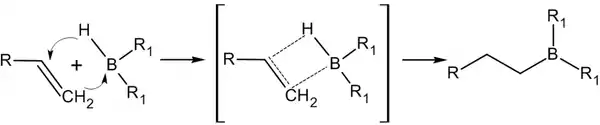
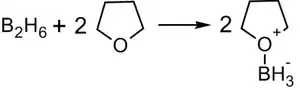
В синтезе используется, обычно, не собственно диборан, а его донорно-акцептоный комплекс с простым эфиром; а сам диборан получают реакцией борогидрида натрия с трифторидом бора в среде тетрагидрофурана[14]:

Алкилбораны легко расщепляются под действием пероксида водорода в щелочной среде, образуя спирты[14]:
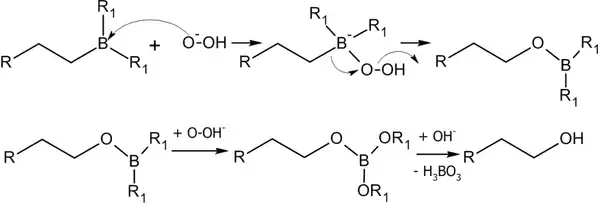
Реакция гидроборирования является реакцией син-присоединения — её результатом становятся цис-аддукты.
Данный метод имеет широкое препаративное значение. Например, алкены с концевой двойной связью дают первичные спирты с выходами 80-90 %. Примеры практического использования метода (в скобках указаны выходы, показывающие соотношение образующихся продуктов)[7]:



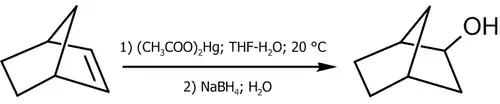
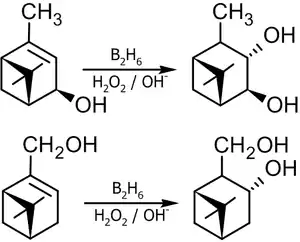
Пример синтеза бициклических терпеновых спиртов[15]:
Для повышения селективности реакции гидроборирования используют замещённые, пространственно затруднённые бораны[16]:
| Тексилборан 2,3-диметилбутил-2-боран | Дисиамилборан бис-(1,2-диметилпропил)боран | 9-ББН 9-борабицикло[3,3,1]нонан | Диизопинокамфеилборан (+)3-дипинанилборан |
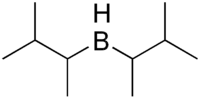 Дисиамилборан | 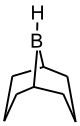
|  Диизопинокамфеилборан |

Использование, например, в реакции со стиролом дисиамилборана (DIAB) повышает выход первичного спирта с 80 % до 98 %[17]:

Высокая селективность указанных выше производных борана позволяет избирательно вступать в реакцию с цис-изомером, находящемся в смеси транс-изомером или гидроборировать одну двойную связь из двух имеющихся в молекуле алкена, например[6]:

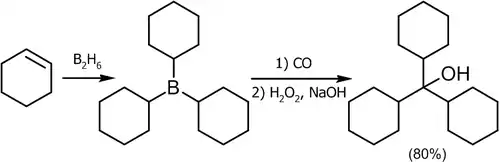
Гидроборирование алкенов с последующим присоединением окиси углерода
Одним из лучших способов получения третичных спиртов является присоединение к окиси углерода к алкилборанам. Реакция легко протекает при обычном давлении и температуре около 125 °C, в качестве растворителя используется диглим[7]:



Этот метод даёт неплохие выходы со многими алкенами[14]:

Если данную реакцию проводить в присутствии водного раствора щёлочи, получаются вторичные спирты[18]:

Гидроформилирование алкенов
Широко используемая в промышленности классическая реакция гидроформилирования алкенов, то есть каталитического присоединение к ним водорода и монооксида углерода с получением на выходе альдегидов[19], может быть проведена таким образом, что продуктом её реакции сразу будут спирты без выделения промежуточных карбонильных соединений Это метод иногда называют восстановительным гидроформилированием.

Катализирует реакцию координационно ненасыщенный гидрокарбонил кобальта, образующийся в ходе реакции[19]:


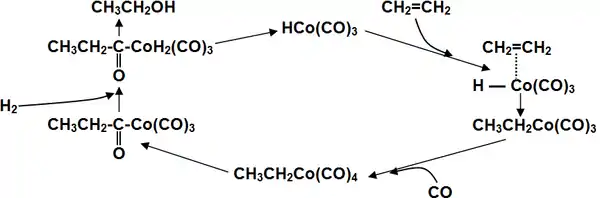
Для получения спиртов в одну стадию в качестве катализаторов используют карбонилы кобальта, модифицированные фосфинами, что помимо более активного гидрирования, позволяет добиться существенно более высокой селективности выхода нормальных продуктов (до 90 %) вследствие стерического эффекта объёмного фосфинового лиганда в переходном состоянии[20].
Реакция гомологизации спиртов
Гомологизация, то есть превращение органического соединения в свой гомолог путём внедрения одной или нескольких метиленовых групп, для спиртов была впервые осуществлена в 1940 году — на основе метанола каталитическим путём под воздействием высокого давления был синтезирован этанол[21]:

Реакция гомологизации по своему механизму близка реакции гидроформилирования алкенов и в настоящее время с помощью модифицированных катализаторов кобальта и рутения и добавления йодид-ионов в качестве промоторов удаётся добиться 90 % выхода по этанолу[21].
Исходный метанол также получают из окиси углерода (катализаторы на основе оксидов меди и цинка, давление 5-10 МПа, температура 250 °C)[21], так что общая схема выглядит следующим образом:

Побочными продуктами реакции в случае синтеза этанола будут ацетальдегид, этилен и диэтиловый эфир.
Реакция Гербе́
Реакция Гербе представляет собой высокотемпературный (200 °C, давление 5—6 МПа) процесс каталитической конденсации первичных алифатических спиртов, не имеющих разветвления в α-положении, по следующей схеме[22]:

В качестве катализаторов используют сложную смесь на основе никеля Ренея, меди, солей железа и других компонентов[23].
Предполагаемый условный механизм реакции[23]:



или

Реакция имеет ограниченное применение как из-за жестких условий её проведения и относительно низкого выхода (как правило, до 70 %), так и образования кислоты и альдегида в качестве побочного продукта[23].
Кислотное расщепление простых эфиров
В лабораторной практике подобный способ получения спиртов крайне редок, так как именно спирты служат исходным компонентом для синтеза простых эфиров. Вместе с тем, если в качестве исходного объекта выбран, скажем природный простой эфир сложной структуры, его лабораторное расщепление до исходного спирта может оказаться востребованным. Кроме того, в некоторых случаях для защиты гидроксильной группы в процессе многоступенчатого синтеза, её могут перевести в эфирную и вводить в реакцию уже простой эфир. По окончании процесса для обратного превращения соединения в спирт может потребоваться расщепление эфира (см. подробнее подраздел «Защита через простые эфиры»).
Обычно реакцию проводят нагреванием эфира и концентрированного раствора бромоводородной или йодоводородной кислоты, при этом расщепление может осуществляться как по механизму SN1, так и SN2[24]:
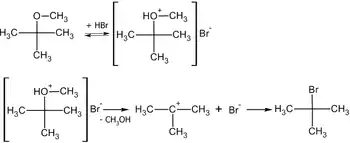 SN1 механизм расщепления простых эфиров |
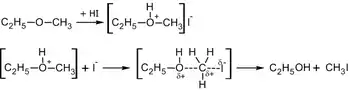 SN2 механизм расщепления простых эфиров |
Если в реакцию вступают несимметричные эфиры, в результате получаются два спирта и два галогенпроизводных, однако если эфир метиловый, продуктом реакции будет спирт и метилиодид или метилбромид[25]:
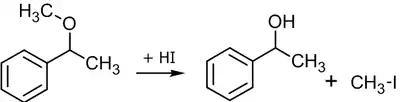
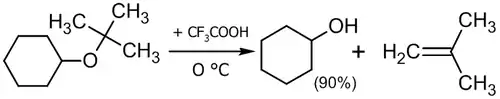
Для расщепления эфиров могут использоваться также кислоты Льюиса: BF3, BCl3, AlCl3 и др[25], а также сильные органические кислоты. Например, расщепление трет-бутилциклогексилового эфира трифторуксуной кислотой происходит по механизму SN1 с образованием циклогексанола и 2-метилпропена[26]:
Перегруппировка Виттига
Простые эфиры под действием фениллития перегруппировываются в спирты (Георг Виттиг, 1942 год):

Реакция представляет собой карбанионную перегруппировку, которая осуществляется через радикальный механизм расщепления-рекомбинации[27]:
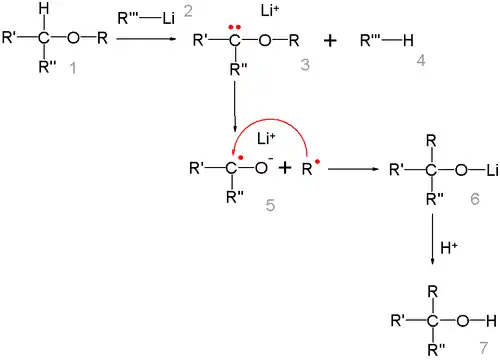
Говоря о стереохимии перегруппировки Виттига, следует отметить, что образование новой C-C связи происходит настолько быстро, что радикал R не успевает инвертироваться, поэтому обычно, реакция протекает с сохранением исходной конфигурации[28]. Изучение перегруппировки на примере β-алкоксиалкилаллиловых эфиров (общий вид: ) показало, что в результате реакции с выходом 14-32 % образовывались син-1,3-диол производные с селективностью 90-95 %[29].

Перегруппировка Виттига может осуществляться не только с помощью алкил- или ариллитиевых соединений (фениллитий, бутиллитий, метиллитий, диэтиламид лития и пр.), но и под действием других сильных оснований; например, следующая реакция протекает в жидком аммиаке в присутствии амида калия (выход 90 %)[30]:
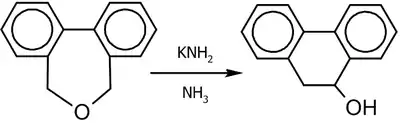
Для аллильно-замещённых субстратов (1,2)-перегруппировка конкурирует с (2,3)-перегруппировкой, которую можно наблюдать практически полностью независимо при низких температурах[27]:
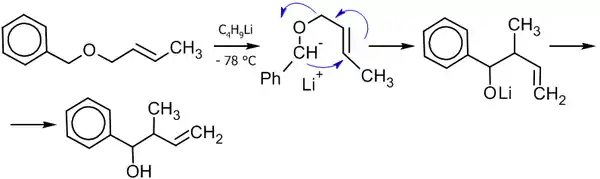
Получение спиртов из альдегидов и кетонов
В данном разделе помимо получения собственно спиртов из альдегидов и кетонов приведены синтезы гидроксикарбонильных соединений (кетоспирты и производные гидроксикарбоновых кислот; смотри подразделы «Альдольная конденсация», «Бензоиновая конденсация», «Реакция Реформатского», «Реакция Иванова»). Это связано с тем, что приведённые реакции являются мощными препаративными методами и широко используются на практике.
Этинилирование карбонильных соединений
Важным методом получения ацетиленовых спиртов является реакция Фаворского, иначе говоря, реакция этинилирования карбонильных соединений:


В реакцию вступают незамещённые алкины (берутся в большом избытке), кетоны и некоторые альдегиды (чаще всего используется формальдегид) в присутствии оснований (KOH или NaNH2 в органическом растворителе) при температуре от −70 до +40 °C, давлении 0,4-0,9 МПа[31].
Механизм данной реакции связан с нуклеофильным присоединеним этинильного карбаниона к карбонильной группе[32]:
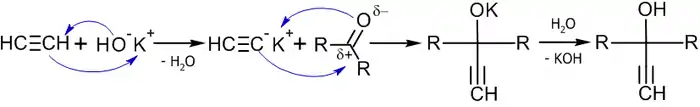
Примеры реакций[33]:


Существуют, по-меньшей мере, две модификации этого метода:
- Реакция Реппе — конденсация алкинов с альдегидами или кетонами в присутствии каталитических количеств ацетиленидов меди, серебра или ртути[34].
- Реакция Нефа — реакция с последующим гидролизом ацетиленидов щелочных металлов с кетонами, включая α,β-непредельные и ароматические карбонильные соединения[35].
Взаимодействие альдегидов с аллилборанами
Современным методом получения аллильных спиртов заданной конфигурации является применение в качестве агента аллилборана, который реагирует с альдегидами в присутствии оснований по следующей схеме[36]:

Реакция была использована, в частности, в качестве базовой для полного синтеза ряда природных соединений и их аналогов, например, феромонов короеда[36]:
| Ипсенол | Ипсдиенол |
| 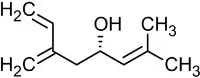 |
Существуют различные методики этой реакции, среди которых:
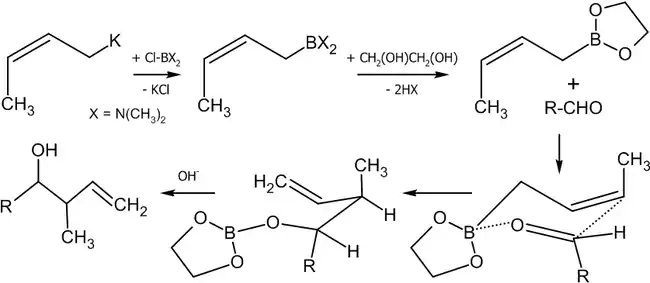
Если в реакцию вступают цис-алкены, в основном, будут образовываться син-продукты присоединения (97 % от общего выхода).
Другим удобным агентом, реагирующим по данной схеме является аллилборный эфир винной кислоты (Рауш, 1985 год)[38].
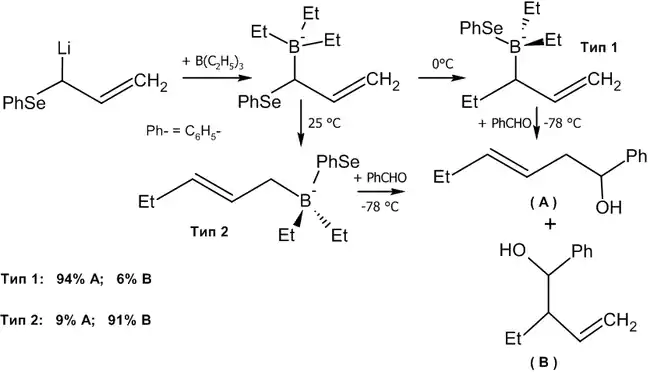
Как видно из схемы, меняя температуру реакции, можно вызвать миграцию атома бора к соседнему углероду, получив тот или иной изомерный спирт.
Реакция Сакураи
Другой способ аллилирования, заключающийся в электрофильном взаимодействии аллилсиланов с различными соединениями в присутствии кислот Льюиса носит название реакции Сакураи. С точки зрения получения спиртов, существуют две модификации подобного синтеза[39]:
- Реакция с карбонильными соединениями с получением вторичных или третичных спиртов:

- Реакция с эпоксидами с получением вторичных спиртов:

В качестве катализаторов реакции могут выступать: TiCl4, BF3, SnCl4 и пр.
Пример практического использования реакции Сакураи[40]:
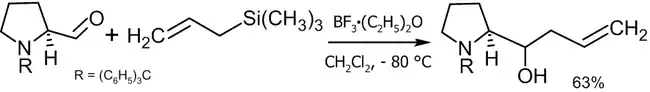
Реакция Бэйлиса-Хиллмана-Морита
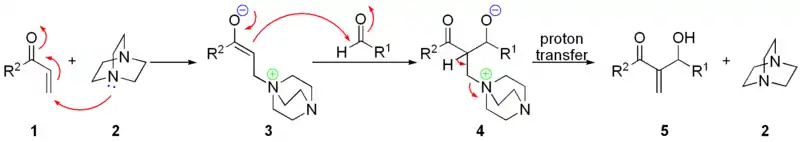
Традиционная реакция Бэйлиса-Хиллмана-Морита представляет собой метод получения аллиловых кетоспиртов взаимодействием альдегидов с метилвинилкетонами или другими активированными алкенами в присутствии третичных фосфинов и каталитических количеств фенола или его производных[41]. Впоследствии реакция была несколько модифицирована: в качестве катализатора стали использовать третичные амины (например: 1,4-диазобицикло[2.2.2]октан или DABCO[42]):
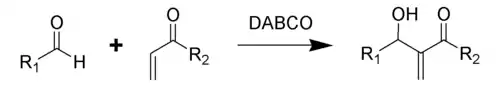
Предположительно, механизм реакции выглядит следующим образом[43]:
Реакция Нозаки-Хияма-Киши
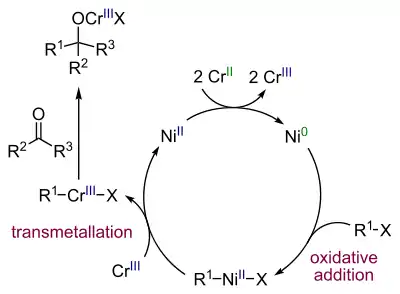
Реакция Нозаки-Хияма-Киши представляет собой современный метод получения спиртов селективным восстановительным сочетанием альдегидов с винил- или аллилгалогенидами (бромиды или йодиды) в присутствии хром-никелевого катализатора[44]:

Каталитический цикл реакции выглядит следующим образом[45]:
Каталитическое сочетание альдегидов с аллиловыми спиртами и их производными
Аналогом синтетического метода, рассмотренного в предыдущем подразделе, является реакция сочетания альдегидов с аллиловыми спиртами и их производными в присутствии катализаторов. В научной литературе описано множество лабораторных методик осуществления подробного синтеза с примененением органических соединений кремния, олова, хрома, лития, рутения, палладия, цинка, титана, циркония и других металлов.
Приведём некоторые характерные примеры использования этого метода на практике:
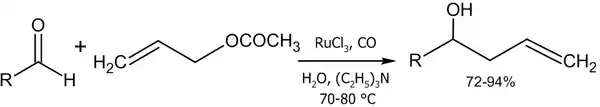
- Реакция аллилацетата с альдегидами в присутствии солей рутения (Denmark S. E., Nguyen S. T., 2009 год)[46]:
- Реакция аллилового спирта с алифатическими альдегидами в присутствии палладиевого катализатора (Masanari Kimura, Masamichi Shimizu, Kazufumi Shibata, Minoru Tazoe, Yoshinao Tamaru, 2003 год)[47]:
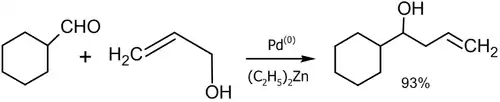
- Реакция аллилтрибутилстанната с альдегидами в присутствии рениевого комплекса (Yutaka Nishiyama, Fujio Kakushoua, Noboru Sonoda, 2004 год)[48]:
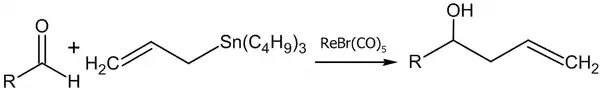
Более подробно о современных методах получения спиртов реакцией аллиловых спиртов и их производных с карбонильными соединениями можно прочитать в монографии: Junzo Otera. Modern Carbonyl Chemistry — Wiley-VCH, Weinheim, 2000—613 Pages — ISBN 978-3-527-29871-6.
Реакция Канниццаро
Реакция Канниццаро представляет собой оксилительно-восстановительное диспропорционирование альдегидов в первичные спирты и карбоновые кислоты под действием оснований[49]:

На первом этапе реакции происходит нуклеофильное присоединение основания (например: гидроксид-аниона) к карбонильному углероду альдегида. Образующийся анион депротонируется (это требует воздействия достаточно сильного основания) с образованием промежуточного дианиона, который затем вступает в реакцию с молекулой альдегида:
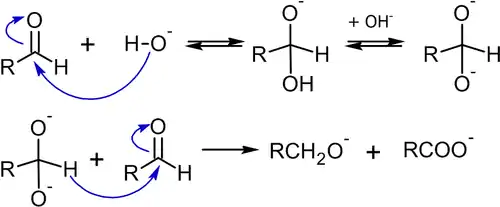
В реакцию Канниццаро вступают альдегиды, не способные к енолизации (не имеющие α-водорода), поскольку для последних преобладающей будет альдольная конденсация. Например, в известном примере с бензальдегидом, выход бензилового спирта может достигать 90 %[50]:

Чаще реакция Канниццаро используется для синтеза ароматических и гетероароматических спиртов[50].
Для повышения реакционного диапазона используемых альдегидов и увеличения выхода спиртов на практике используется перекрестная реакция Канниццаро, то есть использование двух различных альдегидов, причем в качестве альдегида-восстановителя обычно используется формальдегид, который в ходе реакции окисляется в муравьиную кислоту[51]:

В настоящее время существуют более эффективные синтетические методы, поэтому полезность реакции Канниццаро ограничена, как правило, диспропорционированием кетоальдегидов в гидроксикарбоновые кислоты[52]:

Циангидринный синтез
Карбонильные соединения, особенно альдегиды и стерически не затруднённые кетоны, легко вступают в реакции нуклеофильного присоединения c цианистым водородом (нуклеофил CN−) с образованием циангидринов[53]:

Для ароматических кетонов вместо HCN используют цианид диэтилалюминия (C2H5)2AlCN или цианотриметилсилан (CH3)3SiCN, продукт присоединения которого затем гидролизуется до циангидрина[53]:

Далее при необходимости циангидрин легко гидролизуется до гидроксикислоты или восстанавливается в аминоспирт:


Альдольная конденсация
Альдольная конденсация — это одна из старейших реакций органического синтеза (1872 год, Вюрц), в которой две молекулы альдегида или кетона под действием основания или кислоты соединяясь, образуют кетоспирты или альдоли[54]:
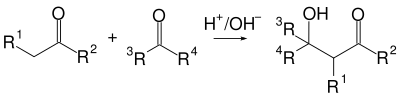
Возможны два механизма этой реакции: щелочной или кислотный, однако с точки зрения синтеза спиртов, последний менее предпочтителен, так как часто реакция не останавливается на стадии спирта а протекает дальше с дегидратацией и образованием непредельных карбонильных соединений (кротоновая конденсация)[54].
Механизм конденсации, происходящий под действием основания, следующий[54]:
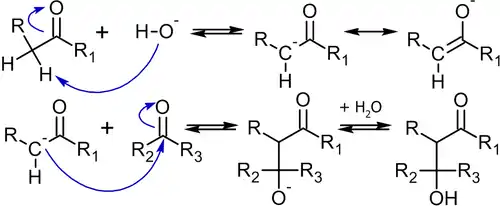
Для проведения конденсации, как видно из её механизма, необходимо, чтобы хотя бы одна из молекул содержала водород в α-положении к карбонильной группе. Обычно, для отрыва этого атома водорода силы гидроксид-иона бывает достаточно, но в отдельных случаях используют и более сильные основания, например — бутиллитий.
Возможны пять комбинаций протекания альдольной конденсации[53]:
- Взаимодействие двух молекул одного альдегида: реакция легко осуществима и приводит к одному продукту, однако из-за электроноакцепторного эффекта альдегидной группы, использование их для синтеза спиртов не эффективно из-за обычно преобладающего расщепления образующегося альдоля до непредельного альдегида, например:

- Взаимодействие двух молекул различных альдегидов: теоретически реакция может привести к четырём разным продуктам, но если один из альдегидов не будет содержать α-карбонильный водород, возможно осуществление только перекрестной реакции;
- Взаимодействие двух молекул одного кетона: реакция сильно смещена влево, поэтому для её проведения либо пользуются специальным оборудованием (аппарат Сокслета), позволяющим фактически удалять из реакционной зоны продукт реакции или использовать в качестве основания особые реагенты (например: нитрид бария);
- Взаимодействие двух молекул разных кетонов: применяется довольно редко и в случаях, когда один из кетонов не содержит α-карбонильный водород;
- Взаимодействие одной молекулы альдегида с одной молекулой кетона: чаще всего в качестве альдегида используют формальдегид, который даёт один продукт конденсации. Другой вариант — использование не самого кетона, а его енольной формы, например в виде литиевой соли или силилового эфира.
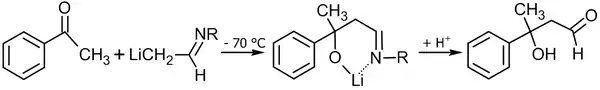
Кроме собственно альдегида, возможно использование имина и диизопропиламида лития в качестве основания[53]:
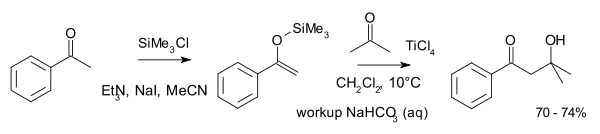
Разновидностью альдольной реакции является реакция Мукаямы, в которой используются силиленольные эфиры:
Бензоиновая конденсация
Бензоиновая конденсация представляет собой обратимое образование из альдегидов (преимущественно — ароматических) под действием цианид-ионов CN− α-оксикетонов (ацилоинов) с общей формулой —СR(ОН)—С(O)О—:

В этой реакции на первом этапе цианид анион вступает в реакцию нуклеофильного присоединения с альдегидом. Перегруппировка образующегося интермедиата в карбанион завершается дальнейшим присоединением второй карбонильной группы также по нуклеофильному механизму. Завершает реакцию переноса протона и отщепление цианидной группы с образованием бензоина в качестве конечного продукта.
Реакция Иванова
Реакция Иванова представляет собой метод получения β-гидроксикарбоновых кислот общей формулой —CR(OH)—CR1(COOH)— из карбонильных соединений и реактивов Иванова: магнийгалогенпроизводных солей арилуксусных кислот (обычно — фенилуксусной кислоты)[55]. Например:

При необходимости оксикислота может быть легко превращена в соответствующий спирт методом декарбоксилирования.
Гидролиз сложных эфиров карбоновых кислот
Гидролиз сложных эфиров карбоновых кислот является типичной реакцией алифатического нуклеофильного замещения, проходящей по следующей условной схеме:

Обычно реакцию проводят при нагревании в щелочной среде. В частности, данный способ является одним из промышленных путей получения глицерина из животных или растительных жиров.
Ацилоиновая конденсация
Ацилоиновая конденсация представляет собой получение α-гидроксикетонов (ацилоинов) восстановлением сложных эфиров металлическим натрием:
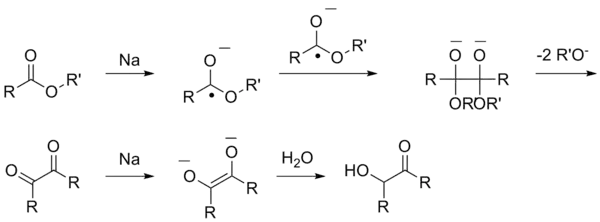
Данная реакция включает в себя несколько стадий, при которых сначала образуются анион-радикалы, а после ряда трансформаций алкоголяты, которые под действием воды переходят в ацилоины.
Декарбонилирование карбоновых кислот
Декарбонилирование (отщепление CO) карбоновых кислот достаточно редкий лабораторный способ получения спиртов, который может быть осуществлён с использованием металлических катализаторов.
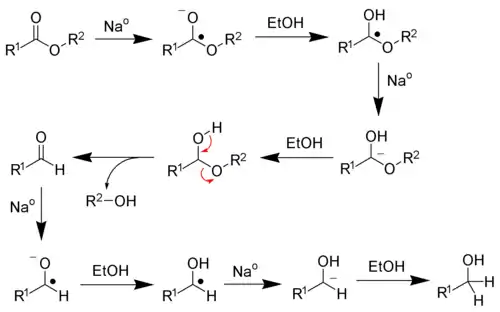
Восстановление карбоновых кислот и сложных эфиров по методу Буво-Блана
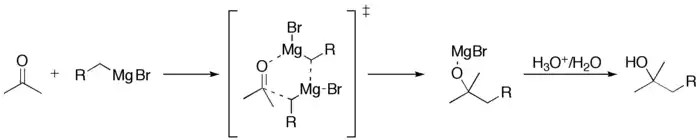
Присоединение реактивов Гриньяра к альдегидам или кетонам
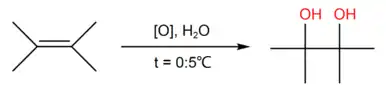
Окисление алкенов
Многоатомные спирты можно получить путём мягкого окисления (по Вагнеру) алкенов - для этого необходимо пропустить их через водный раствор окислителя, например - перманганата калия, - при температуре от 0 до 5 градусов Цельсия:
 Также возможно и получение вторичных спиртов методом жёсткого окисления разветвлённых алкенов (либо подкисленным раствором окислителя, либо оксидом осмия (VIII)) с последующим восстановлением:
Также возможно и получение вторичных спиртов методом жёсткого окисления разветвлённых алкенов (либо подкисленным раствором окислителя, либо оксидом осмия (VIII)) с последующим восстановлением:
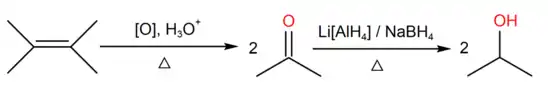
Озонолиз алкенов с последующим восстановлением
Спирт можно получить озонированием алкена с дальнейшей реакцией с сильным восстановителем (тетрагидроборат натрия, тетрадигидроалюминат лития):
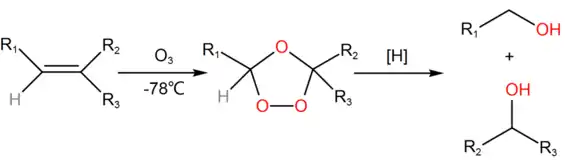
Реакцию озонолиза следует проводить с использованием таких органических растворителей, как хлористый метилен или этилацетат[56].
Реакция Циглера
Является методом синтеза высших спиртов (С8 и выше) при помощи алюминийорганических соединений. Алюминийорганические соединения легко могут быть получены из олефинов в присутствии водорода. Данным методом можно получать чистые первичные спирты:

Окисление реактивов Гриньяра

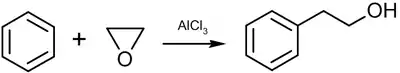
Реакция Фриделя-Крафтса
Получение спиртов в промышленности
Примечания
- Спирты // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 4. — С. 800.
- Курц А.Л., Брусова Г.П., Демьянович В.М. Получение одноатомных спиртов. Одно- и двухатомные спирты, простые эфиры и их сернистые аналоги. ChemNet. Химический факультет МГУ (1999). Дата обращения: 1 сентября 2009.
- Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т. / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — 586-623 с. — ISBN 5-94628-171-2.
- Глава 3.3.3. Нуклеофильное замещение галогена // Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1981. — Т. 1. — С. 660-661.
- Глава 4.1.1. Одноатомные спирты // Общая органическая химия. Кислородсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1982. — Т. 2. — С. 13-118.
- Mарч Дж. Глава 15. Реакции присоединения к кратным связям углерод-углерод // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 3. — С. 132-212.
- Бюлер К., Пирсон Д. Глава 4. Спирты. Б. Реакции присоединения и замещения // Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 1. — С. 213-219.
- Курц А Л., Ливанцов М.В., Ливанцова Л.И. Гидратация алкенов. Алкены (Часть 1). ChemNet. Химический факультет МГУ (1998). Дата обращения: 2 сентября 2009.
- Лебедев. Химия и технология основного органического и нефтехимического синтеза: Учебник для вузов / Пер. с англ. — 4-е изд. перераб. и доп.. — М.: «Химия», 1988. — С. 180-184. — ISBN 5-7245-0008-6.
- Глава 2.2.3. Реакции олефинов // Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1981. — Т. 1. — С. 207-208.
- Carey F.A. Oxymercuration-Demercuration of Alkenes (англ.). Organic chemistry. McGraw-Hill Higher Education. Дата обращения: 2 сентября 2009. Архивировано 23 апреля 2012 года.
- Реутов О.А, Курц А.Л., Бутин К.П. Органическая химия. — М.: Издательство МГУ, 1999. — Т. 1. — С. 385-387. — ISBN 5-211-03054-0.
- The Nobel Prize in Chemistry 1979 (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation. Дата обращения: 3 сентября 2009. Архивировано 22 августа 2011 года.
- Курц А Л., Ливанцов М.В., Ливанцова Л.И. Гидроборирование алкенов (недоступная ссылка). Алкены (Часть 2). ChemNet. Химический факультет МГУ (1998). Дата обращения: 2 сентября 2009. Архивировано 19 декабря 2011 года.
- Кучин А.В., Фролова Л.Л., Пантелеева М.В. Бициклические терпеновые диолы как лиганды для синтеза хиральных катализаторов (pdf) (недоступная ссылка). Англо-русскоязычный общественный химический журнал «Бутлеровские сообщения». Дата обращения: 31 августа 2009. Архивировано 23 апреля 2012 года.
- Борорганические соединения // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 594-603.
- Вацуро К.В., Мищенко Г.Л. 109. Браун (Brown H.C.) // Именные реакции в органичнеской химии. — М.: «Химия», 1976. — С. 76-77.
- Дядченко В.П., Трушков И.В., Брусова Г.П. Синтетические методы органической химии. Части 1-2. — М.: МГУ им. М.В. Ломоносова, 2004. — С. 46.
- Караханов Э.А. Синтез-газ как альтернатива нефти. I. Процесс Фишера-Тропша и оксо-синтез // Соросовский образовательный журнал. — 1997. — № 3. — С. 73-74. (недоступная ссылка)
- Шелдон Р.А. Химические продукты на основе синтез-газа = Chemicals From Synthesis Gas / Под ред. С.М.Локтева. — М.: «Химия», 1987. — С. 92.
- Караханов Э.А. Синтез-газ как альтернатива нефти. II. Метанол и синтезы на его основе // Соросовский образовательный журнал. — 1997. — № 12. — С. 68. (недоступная ссылка)
- Гербе реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 1024-1025.
- Бюлер К., Пирсон Д. Глава 4. Спирты. Ж. Присоединение карбанионов // Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 1. — С. 268-279.
- Травень В.Ф. Глава 18. Простые эфиры. Циклические эфиры // Органическая химия: Учебник для вузов: В 2 т. / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 2. — 97-98 с. — ISBN 5-94628-172-0.
- Mарч Дж. Глава 10. Реакции алифатического нуклеофильного замещения // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 2. — С. 11-240.
- McMurry J. Organic chemistry. — Seven edition. — Thomson, 2008. — P. 604, 658. — ISBN 0-495-11258-5.
- (1,2)-Wittig Rearrangement (англ.). Name Reactions. Organic Chemistry Portal. Дата обращения: 14 сентября 2009. Архивировано 23 апреля 2012 года.
- Бутин К.П. Механизмы органических реакций: достижения и перспективы // Журнал Российского химического общества им. Д.И.Менделеева. — 2001. — Т. XLV, № 2. — С. 32.
- Schreiber S.L., Goulet M. T. Stereochemistry of the 1,2-Wittig Rearangement: A Synthesis of syn-1,3-diol Monoethers (англ.) // Tetrahedron Letters. — 1987. — Vol. 28, no. 10. — P. 1043-1046.
- Физер Л., Физер М. Реагенты для органического синтеза = Reagents For Organic Synthesis / Под редакцией проф. И.Л.Кнунянца и д.х.н. Р.Г.Костяновского. — М.: «Мир», 1970. — Т. 1. — С. 56-57.
- Фаворского реакции // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 5. — С. 95-96.
- Темкин О. Н. Химия ацетилена. «Ацетиленовое дерево» в органической химии XXI века // Соросовский образовательный журнал. — 2001. — Т. 7, № 6. — С. 39. (недоступная ссылка)
- Вацуро К.В., Мищенко Г.Л. 595. Фаворский // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 411-412.
- Вацуро К.В., Мищенко Г.Л. 506. Реппе (Reppe) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 351.
- Вацуро К.В., Мищенко Г.Л. 424. Неф (Nef) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 296.
- Бубнов Ю.Н. Аллилбораны. Принципы реагирования и применение в органическом синтезе // Вестник Московского университета : Серия 2. Химия. — 2005. — Т. 46, № 3. — С. 140-144.
- Reich I.L. Allylborane Reactions (англ.). Chemistry 842 - Fall 2004 Course Outline. University of Wisconsin. Department of Chemistry. Дата обращения: 15 сентября 2009.
- Ли Дж. Роуш (Roush). Аллилборонат как реагент // Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — С. 306. — ISBN 5-94774-368-X.
- Hosomi-Sakurai Reaction (англ.). Name Reactions. Organic Chemistry Portal. Дата обращения: 20 сентября 2009. Архивировано 23 апреля 2012 года.
- Li J. J., Limberakis C., Pflum D. A. Modern organic synthesis in the laboratory: a collection of standard experimental procedures. — New York: Oxford University Press, Inc, 2007. — P. 139. — ISBN 978-0-19-518798-4.
- Traditional Morita-Baylis-Hillman reaction of aldehydes with methyl vinyl ketone co-catalyzed by triphenylphosphine and nitrophenol (англ.). Abstracts. Organic Chemistry Portal. Дата обращения: 23 сентября 2009. Архивировано 23 апреля 2012 года.
- Baylis-Hillman Reaction (англ.) (недоступная ссылка). Name Reactions. Organic Chemistry Portal. Дата обращения: 23 сентября 2009. Архивировано 21 августа 2011 года.
- Smith A.C. Morita Baylis Hillman Reaction (англ.) (pdf). New Methodology and Synthesis of Natural Product. The University of North Carolina at Chapel Hill. Дата обращения: 23 сентября 2009. Архивировано 23 апреля 2012 года.
- Ли Дж. Нозаки-Хияма-Киши (Nozaki-Hiyama-Kishi). Реакция // Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — С. 249. — ISBN 5-94774-368-X.
- Kallemeyn J. M. The Nozaki-Hiyama-Kishi reaction (англ.) (pdf) (недоступная ссылка). The Department of Chemistry at the University of Illinois at Urbana-Champaign (12 апреля 2002). Дата обращения: 15 сентября 2009. Архивировано 23 апреля 2012 года.
- Catalytic, Nucleophilic Allylation of Aldehydes with Allyl Acetate (англ.). Organic Letters. ACS Publications. Дата обращения: 16 сентября 2009. Архивировано 23 апреля 2012 года.
- Pd-Catalyzed Nucleophilic Alkylation of Aliphatic Aldehydes with Allyl Alcohols: Allyl, 2-Tetrahydrofuryl, and 2-Tetrahydropyranyl Ethers as Useful C3, C4, and C5 Sources (англ.) (недоступная ссылка). Angewandte Chemie. Wiley InterScience. Дата обращения: 16 сентября 2009. Архивировано 23 апреля 2012 года.
- Nishiyama Y., Kakushou F., Sonoda N. Rhenium complex-catalyzed allylation of aldehydes with allyltributylstannane (англ.) // Tetrahedron Letters. — 2005. — Vol. 46, no. 5. — P. 787-789.
- Канниццаро реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 603-604.
- Глава 5.3.7. Реакция Канниццаро // Общая органическая химия. Кислородсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1982. — Т. 2. — С. 737-739.
- Mарч Дж. Глава 19. Реакции окисления и восстановления // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 4. — С. 337-338.
- Cannizzaro Reaction (англ.). Name Reactions. Organic Chemistry Portal. Дата обращения: 23 сентября 2009. Архивировано 23 апреля 2012 года.
- Mарч Дж. Глава 16. Реакции присоединения к кратным связям углерод-гетероатом // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 3. — С. 324-427.
- Альдольная конденсация // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 202-204.
- Иванова реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 344—345.
- Алкены II.Окислительное расщепление алкенов. www.chemnet.ru. Дата обращения: 24 февраля 2021.