Активный центр фермента
В биологии активный центр — это область фермента, где молекулы субстрата связываются и подвергаются химической реакции. Активный центр состоит из аминокислотных остатков, которые образуют временные связи с субстратом (сайт связывания), и остатков, которые катализируют реакцию этого субстрата (каталитический сайт). Хотя активный центр занимает только ~ 10-20 % от объёма фермента[1] :19 он является наиболее важной частью, поскольку он непосредственно катализирует химическую реакцию. Обычно активный центр состоит из трех-четырех аминокислот, в то время как другие аминокислоты в белке необходимы для поддержания его третичной структуры[2].
.png.webp)
Каждый активный центр эволюционно развивался для оптимизации связывания с конкретным субстратом и катализирования конкретной реакции, что приводит к высокой специфичности ферментов. Эта специфичность определяется расположением аминокислот в активном центре и структурой субстратов. Иногда ферментам также необходимо связываться с некоторыми кофакторами для выполнения своей функции. Активный центр обычно представляет собой бороздку или карман у фермента, которые могут располагаться в глубоком туннеле внутри фермента[3] или в межфазных границах у мультимерных ферментов. Активный центр может повторно катализировать реакцию, поскольку его аминокислотные остатки не изменяются в конце реакции (они могут изменяться во время реакции, но регенерируются к её концу)[4]. Этот процесс достигается за счет снижения энергии активации реакции, поэтому большее количество субстратов имеет достаточно энергии для прохождения реакции.
Сайт связывания
.png.webp)
.png.webp)
Обычно молекула фермента имеет только два активных центра, и эти активные центры соответствуют одному конкретному типу субстрата. Активный центр содержит сайт связывания, который связывает субстрат и ориентирует его для катализа. Ориентация субстрата и непосредственная близость между ним и активным центром настолько важны, что в некоторых случаях фермент все ещё может функционировать должным образом, даже если все другие его части мутировали и потеряли функцию[5].
Первоначально взаимодействие между активным центром и субстратом нековалентное и временное. Существует четыре важных типа взаимодействия, которые удерживают субстрат в определённой ориентации и образуют комплекс фермент-субстрат (комплекс ES): водородные связи, Ван-дер-Ваальсовы взаимодействия, гидрофобные взаимодействия и электростатические силовые взаимодействия[6] :148. Распределение заряда на субстрате и активном центре должно быть комплементарным, что означает, что все положительные и отрицательные заряды должны быть нейтрализованы. В противном случае их будет раздвигать отталкивающая сила. Активный центр обычно содержит неполярные аминокислоты, хотя иногда могут встречаться и полярные аминокислоты[2]. Связывание субстрата с сайтом связывания требует по крайней мере трех точек контакта для достижения стерео-, регио- и энантиоселективности. Например, алкогольдегидрогеназа, которая катализирует перенос гидрид- иона от этанола к НАД+, взаимодействует с метильной группой субстрата, гидроксильной группой и про-(R) водородом, который будет отводиться во время реакции[6] :149.
Для того чтобы выполнять свою функцию, ферменты должны принять правильную белковую укладку (нативную укладку) и третичную структуру. Чтобы поддерживать заданную трехмерную структуру, белки полагаются на различные типы взаимодействий между своими аминокислотными остатками. Если этим взаимодействиям препятствуют, например, экстремальные значения pH, высокая температура или высокие концентрации ионов, это приведет к денатурированию фермента и потере его каталитической активности.
Считается, что более плотное соединение между активным центром и молекулой субстрата увеличивает эффективность реакции. Если плотность между активным центром ДНК-полимеразы и её субстратом увеличивается, точность, то есть скорость правильной репликации ДНК, также увеличивается[7]. Большинство ферментов имеют глубоко погруженные активные центры, к которым субстрат может получить доступ только через каналы доступа[3].
Предлагаются три модели того, как ферменты соответствуют своему конкретному субстрату: модель замка и ключа, модель индуцированного соответствия и модель конформационного отбора. Последние два не исключают друг друга: конформационный отбор может сопровождаться изменением формы фермента. Кроме того, белок не может полностью соответствовать ни одной из моделей. Аминокислоты в сайте связывания убиквитина обычно следуют модели индуцированного соответствия, тогда как остальной белок обычно придерживается модели конформационного отбора. Факторы, такие как температура, вероятно, влияют на путь, осуществляемый во время связывания, причем более высокие температуры, по прогнозам, увеличивают важность конформационного отбора и уменьшают важность индуцированного соответствия[8].
Гипотеза «замок и ключ»
Эту концепцию предложил химик XIX века Эмиль Фишер. Он предположил, что активный центр и субстрат представляют собой две стабильные структуры, которые идеально подходят без каких-либо дополнительных изменений, точно так же, как ключ вставляется в замок. Если один субстрат идеально связывается со своим активным центром, взаимодействия между ними будут самыми сильными, что приведет к высокой каталитической эффективности.
Со временем начали проявляться ограничения этой модели. Например, конкурентный ингибитор фермента метилглюкозид может прочно связываться с активным центром 4-альфа-глюканотрансферазы и идеально в него вписываться. Однако 4-альфа-глюканотрансфераза не активна в отношении метилглюкозида, и перенос гликозила не происходит. Гипотеза замка и ключа не может этого объяснить, поскольку она предсказывает высокую эффективность переноса метилглюкозид-гликозила из-за его прочного связывания. Помимо конкурентного ингибирования, эта теория не может объяснить механизм действия неконкурентных ингибиторов, поскольку они не связываются с активным центром, но, тем не менее, влияют на каталитическую активность[9].
Гипотеза индуцированной подгонки
Теория связывания фермента с субстратом Дэниела Кошланда состоит в том, что активный центр и связывающая часть субстрата не совсем комплементарны[10]. Модель индуцированной подгонки является развитием модели с замком и ключом и предполагает, что активный сайт является гибким и меняет форму до тех пор, пока субстрат не будет полностью связан. Эта модель похожа на человека, надевающего перчатку: перчатка меняет форму, чтобы соответствовать руке. Фермент изначально имеет конформацию, которая привлекает его субстрат. Поверхность фермента гибкая, и только правильный катализатор может вызвать взаимодействие, ведущее к катализу. При связывании субстрата могут происходить конформационные изменения. После того, как продукты реакции отойдут от фермента, активный центр вернется к исходной форме. Эта гипотеза подтверждается наблюдением, что весь белковый домен может перемещаться на несколько нанометров во время катализа. Движение белковой поверхности может создавать микроокружение, способствующее катализу[5].
Гипотеза конформационного отбора
Эта модель предполагает, что ферменты существуют во множестве конформаций, только некоторые из которых способны связываться с субстратом. Когда субстрат связан с белком, равновесие в конформационном ансамбле смещается в сторону тех, которые способны связывать лиганды (поскольку ферменты со связанными субстратами удаляются из равновесия между свободными конформациями)[11].
Типы нековалентных взаимодействий
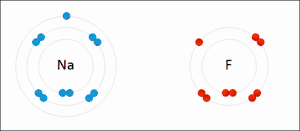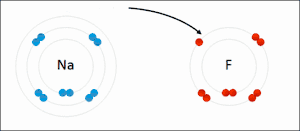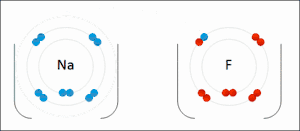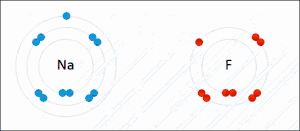
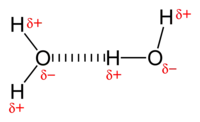
Электростатическое взаимодействие: в водной среде противоположно заряженные группы в боковых цепях аминокислот в активном центре и субстратах притягиваются друг к другу, что называется электростатическим взаимодействием. Например, когда карбоновая кислота (R-COOH) диссоциирует на ионы RCOO- и H+, COO- будет притягивать положительно заряженные группы, такие как протонированная гуанидиновая боковая цепь аргинина.
.png.webp)
Водородная связь: водородная связь — это особый тип диполь-дипольного взаимодействия между частично положительным атомом водорода и частично отрицательным донором электронов, которые содержат пару электронов, таких как кислород, фтор и азот. Прочность водородной связи зависит от химической природы и геометрического расположения каждой группы.
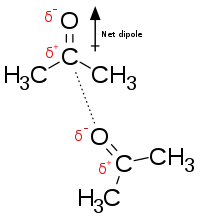
Сила Ван-дер-Ваальса: сила Ван-дер-Ваальса образуется между противоположно заряженными группами из-за временного неравномерного распределения электронов в каждой группе. Если все электроны сосредоточены на одном полюсе группы, этот конец будет отрицательным, а другой конец — положительным. Хотя индивидуальная сила группы мала, общее количество данных взаимодействий между активным центром и субстратом велико, поэтому их сумма может быть значительной.
Гидрофобное взаимодействие: неполярные гидрофобные группы имеют тенденцию агрегироваться вместе в водной среде и пытаться уйти из полярного растворителя. Эти гидрофобные группы обычно имеют длинную углеродную цепь и не реагируют с молекулами воды. При растворении в воде молекула белка сворачивается в шарообразную форму, оставляя гидрофильные группы снаружи, в то время как гидрофобные группы погружаются глубоко в центр.
Каталитический сайт
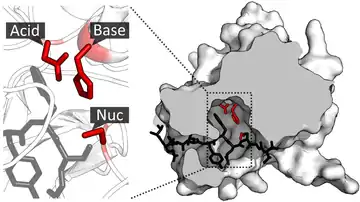
После связывания субстрата с активным центром и его ориентации можно начинать катализ. Аминокислотные остатки каталитического центра обычно очень близки к сайту связывания, и некоторые остатки могут играть двойную роль как в связывании, так и в катализе. Они взаимодействуют с субстратом, чтобы снизить энергию активации реакции и тем самым ускорить её протекание. Снижение энергии активации может происходить с помощью различных механизмов, включая: приближение реагентов, нуклеофильный / электрофильный катализ и кислотно-основной катализ.
Механизмы, участвующие в каталитическом процессе
Приближение реагента
Во время ферментативной каталитической реакции субстрат и активный центр находятся в непосредственной близости. Этот подход преследует разные цели. Во-первых, когда субстраты связываются внутри активного центра, его эффективная концентрация значительно увеличивается, чем в растворе. Это означает, что количество молекул субстрата, участвующих в реакции, также увеличивается. Этот процесс также снижает энергию десольватации, необходимую для протекания реакции. В растворе молекулы субстрата окружены молекулами растворителя, и молекулам фермента требуется энергия для их замены и контакта с субстратом. Поскольку объёмные молекулы могут быть исключены из активного центра, этот выход энергии может быть минимизирован. Далее, активный центр участвует в переориентации субстрата для уменьшения энергии активации реакции. Выравнивание субстрата после связывания блокируется в состоянии высокой энергии и может переходить к следующему этапу. Кроме того, этому связыванию способствует энтропия, поскольку затраты энергии, связанные с реакцией в растворе, в значительной степени устраняются, поскольку растворитель не может попасть в активный центр. В конце концов, активный центр может манипулировать молекулярной орбиталью субстрата в подходящей ориентации для снижения энергии активации[6] :155–8.
Электростатические состояния субстрата и активного центра должны дополнять друг друга. Поляризованная отрицательно заряженная боковая цепь аминокислоты отталкивает незаряженный субстрат. Но если переходное состояние включает образование ионного центра, тогда боковая цепь будет производить благоприятное взаимодействие.
Ковалентный катализ
Многие ферменты, включая сериновую протеазу, цистеиновую протеазу, протеинкиназу и фосфатазу, эволюционировали с образованием временных ковалентных связей между ними и их субстратами, снижая энергию активации и позволяя протекать реакции. Этот процесс можно разделить на 2 этапа: формирование и разрушение. Первый этап является этапом ограничения скорости, в то время как следующий этап необходим для регенерации интактного фермента[6] :158.
Нуклеофильный катализ
Данный процесс включает в себя передачу электронов от нуклеофила фермента субстрату для образования ковалентной связи между ними во время переходного состояния. Сила этого взаимодействия зависит от двух аспектов: способности нуклеофильной группы отдавать электроны и способности электрофила принимать их. Первый в основном зависит от основности (способности отдавать электронные пары) вида, а второй — от его pKa. На обе группы также влияют их химические свойства, такие как поляризуемость, электроотрицательность и потенциал ионизации. Аминокислоты, способные образовывать нуклеофильные реагенты — серин, цистеин, аспартат и глутамин .
Электрофильный катализ
Механизм, лежащий в основе этого процесса, точно такой же, как и нуклеофильный катализ, за исключением того, что теперь аминокислоты в активном центре действуют как электрофилы, а субстраты — как нуклеофилы. Для этой реакции обычно требуются кофакторы, поскольку боковые цепи аминокислот недостаточно сильны для притяжения электронов.
Ионы металлов
Ионы металлов играют несколько ролей во время реакции. Во-первых, они могут связываться с отрицательно заряженными группами субстрата, поэтому они не будут отталкивать электронные пары от нуклеофильных групп активного центра. Они могут притягивать отрицательно заряженные электроны для повышения электрофильности. Ионы металлов также могут служить мостиком между активным центром и субстратом. Наконец, они могут изменить конформационную структуру субстрата в пользу реакции[6] :158.
Кислотно-щелочной катализ
В некоторых реакциях протоны и гидроксид могут непосредственно действовать как кислота и основание при специфическом кислотном и специфическом щелочном катализе. Но чаще группы в субстрате и активном центре действуют как кислота и основание Бренстеда — Лоури. Это называется общей теорией кислоты и общей теории оснований. Самый простой способ различить их — проверить, определяется ли скорость реакции концентрациями общей кислоты и основания. Если «да», то реакция носит общий характер. Поскольку большинство ферментов имеют оптимальный pH от 6 до 7, аминокислоты в боковой цепи обычно имеют pKa 4 ~ 10. Кандидаты включают аспартат, глутамат, гистидин, цистеин. Эти кислоты и основания могут стабилизировать нуклеофил или электрофил, образующийся во время катализа, обеспечивая положительные и отрицательные заряды[6] :164–70.
Конформационное искажение
Количественные исследования ферментативных реакций часто обнаруживали, что ускорение скорости химической реакции не может быть полностью объяснено существующими теориями, такими как приближение, кислотно-основной катализ и электрофильный / нуклеофильный катализ. И здесь возникает очевидный парадокс: в обратимой ферментативной реакции, если активный центр идеально подходит для субстратов, обратная реакция будет замедлена, поскольку продукты не могут идеально вписаться в активный центр. Таким образом, было введено понятие «конформационного искажения» и утверждается, что и активный сайт, и субстрат могут подвергаться конформационным изменениям, для того чтобы соответствовать друг другу все время[6] :170–5.
Предварительно организованная комплементарность активного сайта переходному состоянию
Эта теория немного похожа на теорию замка и ключа, но в ней активный сайт предварительно «запрограммирован» для идеального связывания с субстратом в переходном состоянии, а не в основном состоянии. Формирование переходного состояния в растворе требует большого количества энергии для перемещения молекул растворителя, и реакция замедляется. Таким образом, активный центр может замещать молекулы растворителя и окружать субстраты, чтобы минимизировать контрпродуктивный эффект, вызываемый раствором. Наличие заряженных групп в активном центре будет привлекать субстраты и обеспечивать электростатическую комплементарность[6] :176–8.
Примеры механизмов ферментативного катализа
В действительности, большинство ферментных механизмов включает комбинацию нескольких различных типов катализа.
Глутатионредуктаза
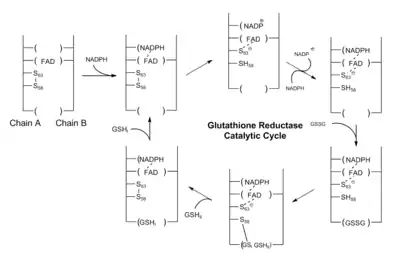
Роль глутатиона (GSH) заключается в удалении накопленных активных форм кислорода, которые могут повреждать клетки. Во время этого процесса его тиоловая боковая цепь окисляется, и две молекулы глутатиона соединяются дисульфидной связью с образованием димера (GSSG). Чтобы регенерировать глутатион, необходимо разорвать дисульфидную связь. В клетках человека это осуществляется глутатионредуктазой (GR).
Глутатионредуктаза представляет собой димер, содержащий две идентичные субъединицы. В качестве кофакторов требуется один НАДФ и один ФАД. Активный сайт находится в связке между двумя субъединицами. НАДФH участвует в генерации ФАДH-. В активном центре есть два остатка цистеина помимо кофактора ФАД, которые используются для разрыва дисульфидной связи во время каталитической реакции. НАДФН связывается тремя положительно заряженными остатками: Arg-218, His-219 и Arg-224.
Каталитический процесс начинается, когда ФАД восстанавливается НАДФН, чтобы принять один электрон, становясь ФАДH-. Затем он атакует дисульфидную связь между 2 остатками цистеина, образуя одну SH связь и одну S- группу. Эта S- группа будет действовать как нуклеофил, чтобы атаковать дисульфидную связь в окисленном глутатионе (GSSG), разрывая её и образуя комплекс цистеин-SG. Первый SG- анион высвобождается, а затем получает один протон от соседней группы SH от первого мономера глутатиона. Затем соседняя S- группа атакует дисульфидную связь в комплексе цистеин-SG и высвобождает второй SG- анион. Он получает один протон в растворе и образует второй мономер глутатиона[1] :137–9.
Химотрипсин
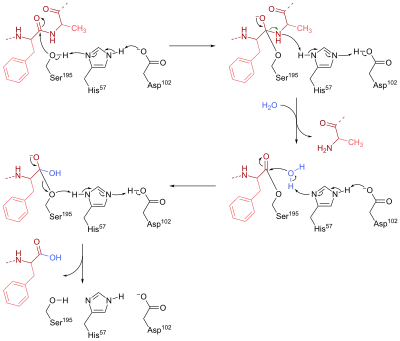
Химотрипсин — это сериновая эндопептидаза, которая присутствует в соке поджелудочной железы и помогает гидролизу белков и пептидов[1] :84–6. Он катализирует гидролиз пептидных связей в L-изомерах тирозина, фенилаланина и триптофана. В активном центре этого фермента три аминокислотных остатка работают вместе, образуя каталитическую триаду, составляющую каталитический центр. В химотрипсине этими остатками являются Ser-195, His-57 и Asp-102.
Механизм действия химотрипсина можно разделить на две фазы. Во-первых, Ser-195 нуклеофильно атакует углерод пептидной связи в субстрате с образованием тетраэдрического промежуточного соединения. Нуклеофильность Ser-195 усиливается His-57, который отрывает протон от Ser-195 и, в свою очередь, стабилизируется отрицательно заряженной карбоксилатной группой (RCOO-) в Asp-102. Кроме того, промежуточный тетраэдрический оксианион, образующийся на этой стадии, стабилизируется водородными связями из Ser-195 и Gly-193.
На второй стадии группа R’NH протонируется His-57 с образованием R’NH2 и оставляет промежуточное соединение, оставляя ацилированный Ser-195. His-57 затем снова действует как основание, чтобы оторвать один протон от молекулы воды. Образовавшийся гидроксид-анион нуклеофильно атакует комплекс ацил-фермент с образованием второго тетраэдрического оксианионного промежуточного соединения, которое снова стабилизируется Н-связями. В конце концов, Ser-195 покидает тетраэдрический промежуточный продукт, разрывая связь CO, которая соединяла фермент с пептидным субстратом. Протон передается на Ser-195 через His-57, так что все три аминокислоты возвращаются в свое исходное состояние.
Отсоединение субстрата
На отсоединение субстрата влияют различные факторы. Лиганды большего размера обычно дольше остаются в активном центре[12] как и лиганды с более вращаемыми связями (хотя это может быть побочным эффектом размера)[13]. Когда растворитель исключен из активного центра, менее «гибкие» белки дольше остаются в активном центре. Большое количество водородных связей, экранированных от растворителя, также замедляют расщепление[12].
Кофакторы
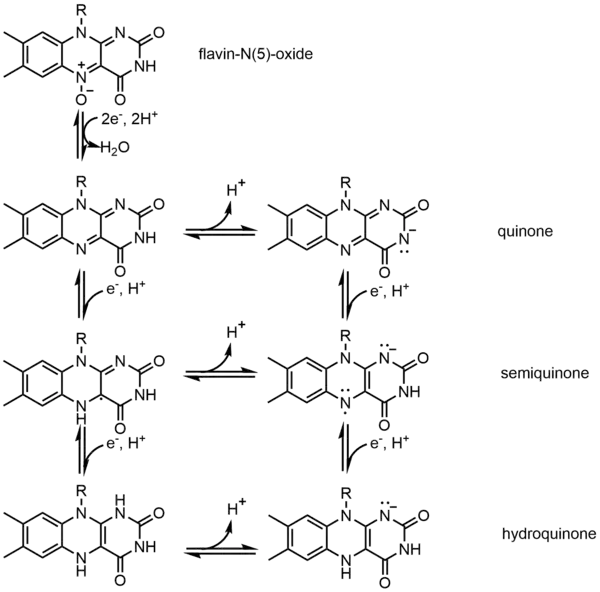
Ферменты могут использовать кофакторы в качестве «вспомогательных молекул». Коферменты относятся к тем небелковым молекулам, которые связываются с ферментами, чтобы помочь им выполнять свою работу. В основном они связаны с активным центром нековалентными связями, такими как водородная связь или гидрофобное взаимодействие. Но иногда между ними также может образовываться ковалентная связь. Например, гем в цитохроме С связан с белком через тиоэфирную связь. В некоторых случаях коферменты могут оставлять ферменты после завершения реакции. В противном случае они навсегда связываются с ферментом[6] :69. Коэнзим — это широкое понятие, которое включает ионы металлов, различные витамины и АТФ . Если ферменту нужен кофермент для работы сам по себе, он называется апоферментом. Фактически, он сам по себе не может должным образом катализировать реакции. Только когда его кофактор входит и связывается с активным центром с образованием холофермента, он работает правильно.
Одним из примеров кофермента является Флавин. Он содержит отдельную конъюгированную изоаллоксазиновую кольцевую систему. Флавин имеет несколько окислительно-восстановительных состояний и может использоваться в процессах, связанных с переносом одного или двух электронов. Он может действовать как акцептор электронов в реакции, такой как окисление НАД до НАДН, принимать два электрона и образовывать 1,5-дигидрофлавин. С другой стороны, он может образовывать семихинон (свободный радикал), принимая один электрон, а затем превращаться в полностью восстановленную форму путем добавления дополнительного электрона. Это свойство позволяет использовать его в процессе одноэлектронного окисления.
Ингибиторы
Ингибиторы нарушают взаимодействие между ферментом и субстратом, замедляя скорость реакции. Существуют разные типы ингибиторов, включая как обратимые, так и необратимые формы.
Конкурентные ингибиторы — это ингибиторы, нацеленные только на свободные молекулы ферментов. Они конкурируют с субстратами за свободный акцептор фермента и их влияние может быть преодолено путем увеличения концентрации субстрата. У них есть два механизма действия. Конкурентные ингибиторы обычно имеют структурное сходство с субстратами и / или комплексом ES. В результате они могут вписаться в активный центр и инициировать взаимодействия, чтобы заполнить пространство фермента и заблокировать проникновение субстратов. Они также могут вызывать временные конформационные изменения в активном центре, чтобы субстраты не могли идеально с ним соответствовать. Через короткий промежуток времени конкурентные ингибиторы отпадут и оставят фермент нетронутым.
Ингибиторы классифицируются как неконкурентные ингибиторы, если они связывают как свободный фермент, так и комплекс ES. Поскольку они не конкурируют с субстратами за активный центр, их нельзя преодолеть простым увеличением концентрации субстрата. Обычно они связываются с другим сайтом фермента и изменяют трехмерную структуру активного центра, чтобы блокировать проникновение или выход субстратов из фермента.
Необратимые ингибиторы похожи на конкурентные ингибиторы, поскольку они оба связываются с активным центром. Однако необратимые ингибиторы образуют необратимые ковалентные связи с аминокислотными остатками в активном центре и никогда не уходят. Следовательно, активный центр занят, и субстрат не может проникнуть. Иногда ингибитор уходит, но форма каталитического центра остается изменённой. Эти ингибиторы обычно содержат электрофильные группы, такие как заменители галогенов и эпоксиды. Со временем все больше и больше ферментов связываются необратимыми ингибиторами и перестают функционировать.
| Пример | Связывает активный центр? | Снижает скорость реакции? | |
|---|---|---|---|
| Конкурентоспособный обратимый ингибитор | Ингибиторы протеазы ВИЧ | да | да |
| Неконкурентный обратимый ингибитор | Тяжелые металлы, такие как свинец и ртуть | Нет | да |
| Необратимый ингибитор | Цианид | да | да |
Примеры конкурентных и необратимых ингибиторов ферментов
Конкурентный ингибитор: ингибитор протеазы ВИЧ.
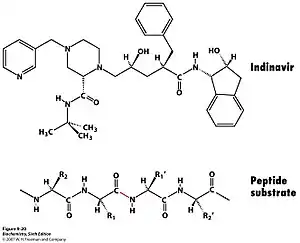
Ингибиторы протеазы ВИЧ используются для лечения пациентов, инфицированных вирусом СПИДа, путем предотвращения репликации его ДНК. Протеаза ВИЧ используется вирусом для расщепления полипротеина Gag-Pol на 3 более мелких белка, которые отвечают за сборку, упаковку и созревание вириона. Этот фермент нацелен на конкретный сайт расщепления фенилаланином и пролином в белке-мишени[14]. Если протеаза ВИЧ выключена, частица вириона потеряет функцию и не сможет инфицировать пациентов. Поскольку он необходим для репликации вируса и отсутствует у здорового человека, он является идеальной мишенью для разработки лекарств.
Протеаза ВИЧ принадлежит к семейству аспарагиновых протеаз и имеет аналогичный механизм. Во-первых, остаток аспартата активирует молекулу воды и превращает её в нуклеофил. Затем он атакует карбонильную группу внутри пептидной связи (NH-CO) с образованием тетраэдрического промежуточного соединения. Атом азота в промежуточном продукте получает протон, образуя амидную группу, и последующая перегруппировка приводит к разрыву связи между ним и промежуточным продуктом и образует два продукта[15].
Ингибиторы обычно содержат негидролизуемые гидроксиэтиленовые или гидроксиэтиламиновые группы, имитирующие тетраэдрический промежуточный продукт. Поскольку они имеют схожую структуру и электростатическое расположение с переходным состоянием субстратов, они все ещё могут помещаться в активный центр, но не могут быть разрушены, поэтому гидролиз не может происходить.
Неконкурентный ингибитор: Стрихнин.
Стрихнин — это нейротоксин, который вызывает смерть, поражая нервы, которые контролируют сокращение мышц и вызывают затруднение дыхания. Импульс передается между синапсами через нейромедиатор, называемый ацетилхолином. Он попадает в синапс между нервными клетками и связывается с рецепторами постсинаптической клетки. Затем генерируется потенциал действия, который передается через постсинаптическую клетку, чтобы начать новый цикл.
Глицин может подавлять активность рецепторов нейромедиаторов, поэтому для запуска потенциала действия требуется большее количество ацетилхолинэстеразы. Это гарантирует, что генерация нервных импульсов строго контролируется. Однако этот контроль нарушается при добавлении стрихнина. Он подавляет рецепторы глицина (хлоридный канал), и гораздо более низкий уровень концентрации нейромедиатора может вызвать потенциал действия. Нервы теперь постоянно передают сигналы и вызывают чрезмерное сокращение мышц, что приводит к удушью и смерти[16].
Необратимый ингибитор: диизопропилфторфосфат.
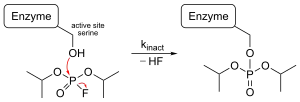
Диизопропилфторфосфат (DIFP) — необратимый ингибитор, блокирующий действие сериновой протеазы. Когда он связывается с ферментом, происходит реакция нуклеофильного замещения с высвобождением одной молекулы фтороводорода. Группа ОН в активном центре действует как нуклеофил, атакуя фосфор в DIFP, образуя тетраэдрический промежуточный продукт и высвобождая протон. Затем связь PF разрывается, один электрон переносится на атом F, и он оставляет промежуточное соединение в виде F- аниона. Он соединяется с протоном в растворе с образованием одной молекулы HF. Между активным центром и DIFP образуется ковалентная связь, поэтому боковая цепь серина больше не доступна для субстрата[17].
В разработке лекарств
Идентификация активных центров имеет решающее значение в процессе открытия лекарств. Трехмерная структура фермента анализируется, чтобы определить аминокислотные остатки активного центра и разработать лекарства, которые могут в них поместиться. Протеолитические ферменты являются мишенями для некоторых лекарств, таких как ингибиторы протеазы, которые включают лекарства против СПИДа и гипертонии[18]. Данные ингибиторы протеаз связываются с активным сайтом фермента и блокируют взаимодействие с природными субстратами[19]. Важным фактором при разработке лекарств является сила связывания между активным центром и ингибитором фермента[20]. Если фермент, обнаруженный в бактериях, значительно отличается от фермента человека, тогда можно разработать ингибитор против этой конкретной бактерии, не нанося вреда ферменту человека. Если один вид ферментов присутствует только в одном виде организмов, его ингибитор можно использовать для их уничтожения.
Активные центры могут быть картированы, чтобы помочь в разработке новых лекарств, таких как ингибиторы ферментов. Это включает в себя описание размера активного центра, количества и свойств подсайтов, таких как детали связывающего взаимодействия[18]. Однако современная технология баз данных под названием CPASS (Сравнение структур протеиновых активных центров) позволяет более детально сравнивать активные центры и находить структурное сходство с помощью программного обеспечения[21].
Применение ингибиторов ферментов
| Пример | Механизм действия | |
|---|---|---|
| Антибактериальное средство | Пенициллин | Стенка бактериальной клетки состоит из пептидогликана. Во время роста бактерий существующее сшивание пептидогликанового волокна нарушается, поэтому новый мономер клеточной стенки может быть интегрирован в клеточную стенку. Пенициллин действует путем ингибирования транспептидазы, которая необходима для образования поперечных связей, поэтому клеточная стенка ослабляется и разрывается из-за тургорного давления. |
| Противогрибковое средство | Азол | Эргостерин — это стерол, который образует поверхностную мембрану грибов. Азол может подавлять его биосинтез, ингибируя ланостерол-14-альфа-деметилазу, поэтому новый эргостерин не вырабатывается, а вредный 14-альфа-ланостерин накапливается в клетке. Кроме того, азол может генерировать активные формы кислорода. |
| Противовирусное средство | Саквинавир | Протеаза ВИЧ необходима для расщепления полипротеина Gag-Pol на 3 отдельных белка, чтобы они могли нормально функционировать и запускать процесс упаковки вируса. Ингибиторы протеазы ВИЧ, такие как саквинавир, подавляют его, поэтому новая зрелая вирусная частица не может быть собрана. |
| Инсектициды | Физостигмин | В нервной системе животных ацетилхолинэстераза необходима для расщепления нейромедиатора ацетилхолина на ацетат и холин. Физостигмин связывается с его активным центром и подавляет его, поэтому импульсный сигнал не может передаваться по нервам. Это приводит к гибели насекомых, поскольку они теряют контроль над работой мышц и сердца. |
| Гербициды | Циклогександион | Циклогександион нацелен на ацетил-КоА-карбоксилазу, которая участвует в первом этапе синтеза жира: АТФ-зависимое карбоксилирование ацетил-КоА до малонил-КоА. Липиды играют важную роль в составе клеточной мембраны. |
Аллостерические сайты
Аллостерический сайт — это сайт на ферменте, не связанный с его активным центром, который может связывать эффекторную молекулу. Это взаимодействие — ещё один механизм регуляции ферментов. Аллостерическая модификация обычно происходит в белках с более чем одной субъединицей. Аллостерические взаимодействия часто присутствуют в метаболических путях и полезны тем, что позволяют одной стадии реакции регулировать другую стадию[19]. Они позволяют ферменту иметь ряд дополнительных молекулярных взаимодействий, помимо высокоспецифичного активного центра[19].
Примечания
- Introduction to Enzyme and Coenzyme Chemistry. — 2nd. — Blackwell Publishing Limited, 2004. — ISBN 9781405114523. Архивная копия от 22 марта 2018 на Wayback Machine
- Enzyme Technology. — I K International Publishing House, 2009. — ISBN 9789380026053.
- “Anatomy of Enzyme Channels”. BMC Bioinformatics. 15: 379. 2014. DOI:10.1186/s12859-014-0379-x. PMID 25403510.
- Essential cell biology. — 3rd ed. — New York: Garland Science, 2010. — xx, 845 pages (various pagings) с. — ISBN 978-0-8153-4129-1, 0-8153-4129-6, 978-0-8153-4130-7, 0-8153-4130-X.
- “How Enzymes Work”. Science. 320 (5882): 1428—1429. 2008. DOI:10.1126/science.1159747. PMID 18556536.
- Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. — 2nd. — Wiley-Blackwell, 2000. — ISBN 9780471359296.
- “Active site tightness and substrate fit in DNA replication”. Annual Review of Biochemistry. 71: 191—219. 1984. DOI:10.1146/annurev.biochem.71.110601.135453. PMID 12045095.
- Csermely, Peter (2010). “Induced fit, conformational selection and independent dynamic segments: an extended view of binding events”. Trends in Biochemical Sciences. 35 (10): 539—546. DOI:10.1016/j.tibs.2010.04.009. ISSN 0968-0004. PMID 20541943.
- “The Key–Lock Theory and the Induced Fit Theory”. Angewandte Chemie International Edition. 33 (2324): 2375—2378. 1995. DOI:10.1002/anie.199423751.
- “Enzymes with lid-gated active sites must operate by an induced fit mechanism instead of conformational selection”. Proceedings of the National Academy of Sciences of the United States of America. 105 (37): 13829—13834. 2008. DOI:10.1073/pnas.0805364105. PMID 18772387.
- Evaluation of Enzyme Inhibitors in Drug Discovery. — 2013. — P. 287–344. — ISBN 978-1-118-54039-8.
- Pan, Albert C. (2013). “Molecular determinants of drug–receptor binding kinetics”. Drug Discovery Today. 18 (13—14): 667—673. DOI:10.1016/j.drudis.2013.02.007. ISSN 1359-6446. PMID 23454741.
- Miller, Duncan C. (2012). “Investigation of the effect of molecular properties on the binding kinetics of a ligand to its biological target”. MedChemComm. 3 (4): 449—452. DOI:10.1039/c2md00270a. ISSN 2040-2503.
- “HIV-protease inhibitors”. The New England Journal of Medicine. 338 (18): 1281—1292. 1998. DOI:10.1056/NEJM199804303381808. PMID 9562584.
- “HIV-1 protease: mechanism and drug discovery”. Organic & Biomolecular Chemistry. 1 (1): 5—14. 2003. DOI:10.1039/B208248A. PMID 12929379.
- “Cytoprotection by inhibition of chloride channels: The mechanism of action of glycine and strychnine”. Life Sciences. 53 (15): 1211—1215. 1993. DOI:10.1016/0024-3205(93)90539-F. PMID 8412478.
- “Mode of inhibition of chymotrypsin by diisopropyl fluorophosphate; introduction of phosphorus”. The Journal of Biological Chemistry. 179 (1): 201—204. 1949. PMID 18119235.
- “Mapping of the active site of proteases in the 1960s and rational design of inhibitors/drugs in the 1990s”. Current Protein & Peptide Science. 6 (6): 501—512. 2005. DOI:10.2174/138920305774933286. PMID 16381600.
- “Allosteric drugs: thinking outside the active-site box”. Chemistry & Biology. 7 (5): 103—107. 2000. DOI:10.1016/S1074-5521(00)00115-0. PMID 10801477.
- “Structure-Based Drug Design: Exploring the Proper Filling of Apolar Pockets at Enzyme Active Sites”. Journal of Organic Chemistry. 73 (12): 4345—4361. 2008. DOI:10.1021/jo800527n. PMID 18510366.
- “Comparison of protein active site structures for functional annotation of proteins and drug design”. Proteins. 65: 124—135. 2006. DOI:10.1002/prot.21092. PMID 16862592.
Дальнейшее чтение
- Alan Fersht, Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, 1998. WH Freeman, 1998.ISBN 0-7167-3268-8 ISBN 0-7167-3268-8
- Bugg, T. Introduction to Enzyme and Coenzyme Chemistry. (2nd edition), Blackwell Publishing Limited, 2004 г.ISBN 1-4051-1452-5 ISBN 1-4051-1452-5 .