Глюкокиназа
Глюкокиназа (Шифр КФ 2.7.1.2) представляет собой фермент, который способствует фосфорилированию глюкозы до глюкозо-6-фосфата. Глюкокиназа содержится в клетках печени и поджелудочной железы человека и большинства других позвоночных. В каждом из этих органов он играет важную роль в регулировании углеводного обмена, действуя как датчик глюкозы, вызывая сдвиги в метаболизме или функцях клеток в ответ на повышение или понижение уровня глюкозы, например, после еды или при голодании. Мутации гена этого фермента могут вызывать необычные формы диабета или гипогликемии.
| Глюкокиназа | |
|---|---|
 | |
| Идентификаторы | |
| Шифр КФ | 2.7.1.2 |
| Номер CAS | 9001-36-9 |
| Базы ферментов | |
| IntEnz | IntEnz view |
| BRENDA | BRENDA entry |
| ExPASy | NiceZyme view |
| MetaCyc | metabolic pathway |
| KEGG | KEGG entry |
| PRIAM | profile |
| PDB structures | RCSB PDB PDBe PDBj PDBsum |
| Gene Ontology | AmiGO • EGO |
| Поиск | |
| PMC | статьи |
| PubMed | статьи |
| NCBI | NCBI proteins |
| CAS | 9001-36-9 |
Глюкокиназа (ГК) представляет собой изофермент гексокиназы, гомологично связанный по крайней мере с тремя другими гексокиназами[1]. Все гексокиназы могут опосредовать фосфорилирование глюкозы до глюкозо-6-фосфата (Г6Ф), что является первой стадией как синтеза гликогена, так и гликолиза. Однако глюкокиназа кодируется отдельным геном, и её отличительные кинетические свойства позволяют ей выполнять другой набор функций. Глюкокиназа имеет более низкое сродство к глюкозе, чем другие гексокиназы, и её активность локализована в нескольких типах клеток, в результате чего остальные три гексокиназы являются более важными факторами подготовки глюкозы для гликолиза и синтеза гликогена для большинства тканей и органов. Из-за этого пониженного сродства активность глюкокиназы в обычных физиологических условиях существенно варьируется в зависимости от концентрации глюкозы[2].
Номенклатура
Альтернативные названия этого фермента: человеческая гексокиназа IV, гексокиназа D и АТФ:D-гексоза 6-фосфотрансфераза, ЕС 2.7.1.1 (ранее 2.7.1.2). Общее название глюкокиназа происходит от её относительной специфичности к глюкозе в физиологических условиях.
Некоторые биохимики утверждают, что следует отказаться от названия глюкокиназа как вводящего в заблуждение, поскольку этот фермент может фосфорилировать другие гексозы в правильных условиях, а у бактерий есть отдаленно родственные ферменты с более абсолютной специфичностью для глюкозы, которые лучше заслуживают названия и EC 2.7. 1.2[2][3]. Тем не менее, название глюкокиназа остается предпочтительным в контексте медицины и физиологии млекопитающих.
Другая глюкозокиназа млекопитающих, АДФ-специфическая глюкокиназа, была открыта в 2004 году[4] Этот ген отличается и похож на ген примитивных организмов. Он зависит от АДФ, а не от АТФ (что предполагает возможность более эффективного функционирования при гипоксии), а метаболическую роль и важность ещё предстоит выяснить.
Катализ
Субстраты и продукты
Основным физиологическим субстратом глюкокиназы является глюкоза, а наиболее важным продуктом — глюкозо-6-фосфат. Другой необходимый субстрат, из которого получают фосфат, — это аденозинтрифосфат (АТФ), который при удалении фосфата превращается в аденозиндифосфат (АДФ).
Реакция, катализируемая глюкокиназой:

АТФ участвует в реакции в форме комплекса с магнием (Mg) в качестве кофактора. Кроме того, при определённых условиях глюкокиназа, как и другие гексокиназы, может индуцировать фосфорилирование других гексоз (6-ти углеродных сахаров) и подобных молекул. Таким образом, общая реакция глюкокиназы более точно описывается как:[3]
- Гексоза + MgATP2- → Гексоза-PO2-3 + MgATP- + H+
Среди субстратов гексозы есть манноза, фруктоза и глюкозамин, но сродство глюкокиназы к ним требует концентраций, не обнаруживаемых в клетках для значительной активности[5].
Кинетика
Два важных кинетических свойства отличают глюкокиназу от других гексокиназ, что позволяет ей выполнять особую роль в качестве сенсора глюкозы.
- Глюкокиназа имеет более низкое сродство к глюкозе, чем другие гексокиназы. Глюкокиназа изменяет конформацию и/или функцию параллельно с повышением концентрации глюкозы в физиологически важном диапазоне 4-10 ммоль/л (72-180 мг/дл). Она наполовину насыщена при концентрации глюкозы около 8 ммоль/л (144 мг/дл)[6][7].
- Глюкокиназа не ингибируется её продуктом, глюкозо-6-фосфатом[6]. Это позволяет непрерывно выводить сигнал (например, запускать высвобождение инсулина) среди значительных количеств её продукта[7].
Эти две функции позволяют глюкокиназе регулировать метаболический путь, «управляемый поставками». То есть скорость реакции зависит от предложения глюкозы, а не от спроса на конечные продукты.
Ещё одним отличительным свойством глюкокиназы является её умеренная кооперативность с глюкозой с коэффициентом Хилла (nH) около 1,7[7]. Глюкокиназа имеет только один сайт связывания для глюкозы и является единственным мономерным регуляторным ферментом, который, как известно, проявляет кооперативность с субстратом. Постулируется, что природа кооперативности включает «медленный переход» между двумя различными состояниями фермента с разной скоростью активности. Если доминирующее состояние зависит от концентрации глюкозы, оно будет производить очевидную кооперативность, аналогичную наблюдаемой[8].
Из-за этой кооперативности кинетическое взаимодействие глюкокиназы с глюкозой не соответствует классической кинетике Михаэлиса-Ментен. Вместо Km для глюкозы более точно описать уровень половинного насыщения S0,5, который представляет собой концентрацию, при которой фермент является насыщенным и активным на 50 %.
S0,5 и nH экстраполируются в «точку перегиба» кривой, описывающей активность фермента в зависимости от концентрации глюкозы около 4 ммоль/л.[9] Другими словами, при концентрации глюкозы около 72 м/дл, что близко к нижнему пределу нормального диапазона, активность глюкокиназы наиболее чувствительна к небольшим изменениям концентрации глюкозы.
Кинетическая связь с другим субстратом, MgATP, может быть описана классической кинетикой Михаэлиса-Ментен со сродством примерно 0,3-0,4 ммоль/л, что значительно ниже типичной внутриклеточной концентрации 2,5 ммоль/л. Тот факт, что почти всегда имеется избыток доступного АТФ, означает, что концентрация АТФ редко влияет на активность глюкокиназы.
Максимальная удельная активность (kcat, также известная как скорость оборота) глюкокиназы при насыщении обоими субстратами составляет 62 /с.[6]
Оптимум pH глюкокиназы человека был идентифицирован только недавно, и он неожиданно высок и составляет 8,5-8,7[10].
«Минимальная математическая модель» была разработана на основе вышеуказанной кинетической информации для прогнозирования скорости фосфорилирования глюкозы бета-клеток (BGPR) нормальной («дикого типа») глюкокиназы и её известных мутаций. BGPR для глюкокиназы дикого типа составляет около 28 % при концентрации глюкозы 5 ммоль/л, что указывает на то, что фермент работает на 28 % мощности при обычном пороговом уровне глюкозы для запуска высвобождения инсулина.
Механизм
Сульфгидрильные группы нескольких цистеинов окружают сайт связывания глюкозы. Все, кроме Цис-230, необходимы для каталитического процесса, образуя множественные дисульфидные мостики во время взаимодействия с субстратами и регуляторами. По крайней мере, в бета-клетках соотношение активных и неактивных молекул глюкокиназы, по крайней мере, частично определяется балансом окисления сульфгидрильных групп или восстановления дисульфидных мостиков.
Эти сульфгидрильные группы весьма чувствительны к окислительному статусу клеток, что делает глюкокиназу одним из компонентов, наиболее уязвимых к окислительному стрессу, особенно в бета-клетках.
Структура
| Глюкокиназа | |
|---|---|
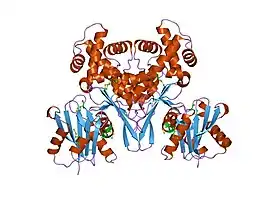 Структура АТФ-зависимой глюкокиназы "Escherichia coli"[11]. | |
| Идентификаторы | |
| Pfam | PF02685 |
| Pfam clan | CL0108 |
| SCOP | 1q18 |
| SUPERFAMILY | 1q18 |
| Доступные структуры белков | |
| Pfam | структуры |
| PDB | RCSB PDB; PDBe; PDBj |
| PDBsum | 3D-модель |
Глюкокиназа — это мономерный белок, состоящий из 465 аминокислот и молекулярной массы около 50 кДа. На поверхности есть по крайней мере две расщелины, одна для активного сайта, связывающего глюкозу и MgATP, а другая для предполагаемого аллостерического активатора, который ещё не идентифицирован[12][13].
Это примерно вдвое меньше, чем у других гексокиназ млекопитающих, которые сохраняют определённую степень димерной структуры. Связывающий АТФ домен является общим с гексокиназами, бактериальными глюкокиназами и другими белками, и общая структура называется укладкой актина .
Генетика
Глюкокиназа человека кодируется геном GCK на хромосоме 7. Этот единственный аутосомный ген имеет 10 экзонов[14][15]. Гены глюкокиназы у других животных гомологичны GCK человека[6][16].
Отличительной особенностью гена является то, что он начинается двумя промоторными участками[17]. Первый экзон с 5'-конца содержит две тканеспецифичные промоторные области. Транскрипция может начинаться с любого промотора (в зависимости от ткани), так что один и тот же ген может продуцировать несколько разные молекулы в печени и в других тканях. Две изоформы глюкокиназы различаются только 13-15 аминокислотами на N-конце молекулы, что дает лишь минимальное различие в структуре. Две изоформы имеют одинаковые кинетические и функциональные характеристики[2].
Первый промотор с 5'-конца, называемый «вышестоящим» или нейроэндокринным промотором, активен в клетках островков поджелудочной железы, нервной ткани и энтероцитах (клетках тонкого кишечника), продуцируя «нейроэндокринную изоформу» глюкокиназы[17]. Второй промотор, «нижележащий» или печеночный промотор, активен в гепатоцитах и направляет продукцию «изоформы печени»[18]. Два промотора имеют небольшую гомологию последовательностей или не имеют её и разделены последовательностью 30 тыс. пар оснований, которая, как ещё не было показано, вызывает какие-либо функциональные различия между изоформами[2]. Два промотора функционально исключают друг друга и регулируются разными наборами регуляторных факторов, так что экспрессия глюкокиназы может регулироваться отдельно в разных типах тканей[2]. Эти два промотора соответствуют двум широким категориям функции глюкокиназы: в печени глюкокиназа действует как ворота для «массовой обработки» доступной глюкозы, в то время как в нейроэндокринных клетках она действует как сенсор, запускающий клеточные реакции, которые влияют на организм: широкий углеводный обмен.
Распределение по системам органов
Глюкокиназа была обнаружена в определённых клетках четырёх типов тканей млекопитающих: печени, поджелудочной железы, тонкой кишки и мозга. Все они играют решающую роль в реагировании на повышение или понижение уровня глюкозы в крови.
- Преобладающими клетками печени являются гепатоциты, и ГК обнаруживается исключительно в этих клетках. Во время переваривания углеводной пищи, когда глюкозы в крови много, а уровень инсулина высок, гепатоциты удаляют глюкозу из крови и хранят её в виде гликогена. После завершения переваривания и всасывания печень производит глюкозу как из не-глюкозных субстратов (глюконеогенез), так и из гликогена (гликогенолиз) и экспортирует её в кровь для поддержания адекватного уровня глюкозы в крови во время голодания. Поскольку активность ГК быстро возрастает по мере повышения концентрации глюкозы, она служит центральным метаболическим переключателем для переключения углеводного обмена в печени между состояниями питания и голодания. Фосфорилирование глюкозы до глюкозо-6-фосфата ГК облегчает хранение глюкозы в виде гликогена и утилизацию путем гликолиза. Отдельный промотор печени позволяет регулировать глюкокиназу в гепатоцитах иначе, чем в нейроэндокринных клетках.
- Нейроэндокринные клетки поджелудочной железы, кишечника и головного мозга имеют некоторые общие аспекты выработки, регуляции и функционирования глюкокиназы[19]. В этом контексте эти ткани в совокупности называются «нейроэндокринными» клетками.
- Бета-клетки и альфа-клетки островков поджелудочной железы
- Бета — клетки выделяют инсулин в ответ на повышение уровня глюкозы. Инсулин позволяет многим типам клеток импортировать и использовать глюкозу, а также сигнализирует печени о необходимости синтеза гликогена. Альфа-клетки вырабатывают меньше глюкагона в ответ на повышение уровня глюкозы и больше глюкагона, если уровень глюкозы в крови низкий. Глюкагон служит сигналом печени для расщепления гликогена и высвобождения глюкозы в кровь. Глюкокиназа в бета-клетках служит датчиком глюкозы, усиливая секрецию инсулина по мере повышения уровня глюкозы в крови.
- В бета-клетках поджелудочной железы глюкокиназа является ключевым ферментом-регулятором. Глюкокиназа очень важна в регуляции секреции инсулина и известна как датчик бета-клеток поджелудочной железы. Мутации в гене, кодирующем глюкокиназу, могут вызывать как гипергликемию, так и гипогликемию из-за её центральной роли в регуляции высвобождения инсулина[20].
- Чувствительные к глюкозе нейроны гипоталамуса
- В ответ на повышение или понижение уровня глюкозы клетки гипоталамуса поляризуются или деполяризуются. Среди нейроэндокринных реакций центральной нервной системы на гипогликемию-активация адренергических реакций вегетативной нервной системы. Глюкокиназа, вероятно, также служит сигналом глюкозы и здесь. Глюкокиназа также была обнаружена в клетках передней доли гипофиза.
- Энтероциты тонкой кишки
- Это наименее изученная из сенсорных систем глюкокиназы. Представляется вероятным, что реакции на поступающую глюкозу во время пищеварения играют роль в инкретиновом усилении секреции инсулина во время еды или в генерации сигналов насыщения от кишечника к мозгу.
- Бета-клетки и альфа-клетки островков поджелудочной железы
Распространение среди животных
Глюкокиназа печени встречается широко, но не повсеместно у позвоночных. Структура гена и аминокислотная последовательность высоко консервативны у большинства млекопитающих (например, глюкокиназа крысы и человека гомологична более чем на 80 %). Однако есть некоторые необычные исключения: например, она не была обнаружена у кошек и летучих мышей, хотя есть у некоторых рептилий, птиц, земноводных и рыб. Происходит ли аналогичное действие глюкокиназы в поджелудочной железе и других органах, ещё не установлено. Было высказано предположение, что присутствие глюкокиназы в печени отражает легкость, с которой углеводы могут быть включены в рацион животных.
Функция и регулирование
Большая часть глюкокиназы у млекопитающих находится в печени, а глюкокиназа обеспечивает примерно 95 % активности гексокиназы в гепатоцитах. Фосфорилирование глюкозы до глюкозо-6-фосфата глюкокиназой является первым этапом как синтеза гликогена, так и гликолиза в печени.
Когда доступно достаточное количество глюкозы, синтез гликогена продолжается на периферии гепатоцитов до тех пор, пока клетки не наполнятся гликогеном. Затем избыток глюкозы все больше превращается в триглицериды для экспорта и хранения в жировой ткани. Активность глюкокиназы в цитоплазме повышается и падает с доступной глюкозой.
Глюкозо-6-фосфат, продукт глюкокиназы, является основным субстратом синтеза гликогена, а глюкокиназа имеет тесную функциональную и регуляторную связь с синтезом гликогена. При максимальной активности глюкокиназа и гликогенсинтаза, по-видимому, расположены в тех же периферических областях цитоплазмы гепатоцитов, в которых происходит синтез гликогена. Поставка глюкозо-6-фосфата влияет на скорость синтеза гликогена не только в качестве основного субстрата, но и путем прямой стимуляции гликогенсинтазы и ингибирования гликогенфосфорилазы .
Активность глюкокиназы может быстро увеличиваться или подавляться в ответ на изменения в поставке глюкозы, обычно возникающие в результате приема пищи и голодания. Регулирование происходит на нескольких уровнях и на нескольких скоростях, и на него влияют многие факторы, которые влияют в основном на два общих механизма:
- Активность глюкокиназы может быть увеличена или уменьшена за считанные минуты под действием регуляторного белка глюкокиназы (GKRP). На действие этого белка влияют небольшие молекулы, такие как глюкоза и фруктоза.
- Количество глюкокиназы можно увеличить за счет синтеза нового белка. Инсулин является основным сигналом для увеличения транскрипции, действуя в основном посредством фактора транскрипции, называемого белком, связывающим регуляторный элемент стерола-1c (SREBP1c), за исключением печени. Это происходит в течение часа после повышения уровня инсулина, как после углеводной еды.
Транскрипционная
Инсулин, действующий через белок −1c, связывающий регуляторный элемент стерола (SREBP1c), считается наиболее важным прямым активатором транскрипции гена глюкокиназы в гепатоцитах. SREBP1c — это базовый трансактиватор типа спираль-петля-спираль-молния (bHLHZ). Трансактиваторы этого класса связываются с последовательностью «Е-бокса» генов ряда регуляторных ферментов. Промотор печени в первом экзоне гена глюкокиназы включает такой E-бокс, который, по-видимому, является основным элементом инсулинового ответа гена в гепатоцитах. Ранее считалось, что SREBP1c должен присутствовать для транскрипции глюкокиназы в гепатоцитах, однако недавно было показано, что транскрипция глюкокиназы осуществляется нормально у мышей с нокаутом SREBP1c. SREBP1c увеличивается в ответ на высокоуглеводную диету, что, как предполагается, является прямым следствием частого повышения уровня инсулина. Повышенную транскрипцию можно обнаружить менее чем через час после воздействия на гепатоциты повышенного уровня инсулина.
Фруктозо-2,6-бисфосфат (Ф2,6БФ2) также стимулирует транскрипцию ГК, по-видимому, посредством Akt2, а не SREBP1c. Неизвестно, является ли этот эффект одним из последующих эффектов активации рецепторов инсулина или не зависит от действия инсулина. Уровни F2,6P2 играют другие усиливающие роли в гликолизе в гепатоцитах. 2 играют другие усиливающие роли в гликолизе в гепатоцитах. Другие факторы трансакции, которые, как предполагается, играют роль в регуляции транскрипции клеток печени, включают:
- Ядерный фактор печени-4-альфа (HNF4α) — это орфанный ядерный рецептор, важный для транскрипции многих генов ферментов углеводного и липидного обмена. Активирует транскрипцию GCK.
- Восходящий стимулирующий фактор 1 (USF1) является ещё одним базовым трансактиватором спиральной молнии (bHLHZ).
- Ядерный фактор печени 6 (HNF6) представляет собой гомеодоменный регулятор транскрипции класса «один разрез». HNF6 также участвует в регуляции транскрипции глюконеогенных ферментов, таких как глюкозо-6-фосфатаза и фосфоенолпируваткарбоксикиназа.
Гормонально-диетическая
Инсулин, безусловно, является наиболее важным из гормонов, которые прямо или косвенно влияют на экспрессию и активность глюкокиназы в печени. Инсулин, по-видимому, влияет как на транскрипцию, так и на активность глюкокиназы множеством прямых и непрямых путей. В то время как повышение уровня глюкозы в воротной вене увеличивает активность глюкокиназы, сопутствующее повышение уровня инсулина усиливает этот эффект за счет индукции синтеза глюкокиназы. Транскрипция глюкокиназы начинает расти в течение часа после повышения уровня инсулина. Транскрипция глюкокиназы становится практически невыявимой при длительном голодании, тяжелой углеводной недостаточности или нелеченном инсулино-дефицитном диабете.
Механизмы, с помощью которых инсулин индуцирует глюкокиназу, могут включать как основные внутриклеточные пути действия инсулина, так и каскад киназ, регулируемых внеклеточными сигналами (ERK 1/2), и каскад фосфоинозитид-3-киназ (PI3-K). Последний может работать через трансактиватор FOXO1.
Однако, как и следовало ожидать, учитывая его антагонистический эффект на синтез гликогена, глюкагон и его внутриклеточный вторичный мессенджер цАМФ подавляют транскрипцию и активность глюкокиназы даже в присутствии инсулина.
Другие гормоны, такие как трийодтиронин (T3), и глюкокортикоиды при определённых обстоятельствах оказывают разрешающее или стимулирующее действие на глюкокиназу. Биотин и ретиноевая кислота увеличивают транскрипцию мРНК GCK, а также активность GK. Жирные кислоты в значительных количествах усиливают активность GK в печени, в то время как длинноцепочечный ацил-КоА ингибирует её.
Печёночная
Глюкокиназа может быть быстро активирована и инактивирована в гепатоцитах новым регуляторным белком (регуляторным белком глюкокиназы — ГКРБ), который поддерживает неактивный резерв ГК, который может быстро стать доступным в ответ на повышение уровня глюкозы в воротной вене[21].
ГКРБ перемещается между ядром и цитоплазмой гепатоцитов и может быть привязан к цитоскелету микрофиламентов. Он образует обратимые комплексы 1:1 с ГК и может перемещать его из цитоплазмы в ядро. Он действует как конкурентный ингибитор глюкозы, так что активность фермента снижается почти до нуля при связывании ГК: комплексы ГКРБ секвестрируются в ядре, в то время как уровни глюкозы и фруктозы низкие. Секвестрация ядра может служить для защиты ГК от деградации цитоплазматическими протеазами. ГК может быстро высвобождаться из ГКРБ в ответ на повышение уровня глюкозы. В отличие от ГК в бета-клетках, ГК в гепатоцитах не связан с митохондриями.
Фруктоза в крошечных (микромолярных) количествах (после фосфорилирования кетогексокиназой до фруктозо-1-фосфата (Ф1Ф)) ускоряет высвобождение ГК из ГКРБ. Эта чувствительность к присутствию небольшого количества фруктозы позволяет ГКРБ, ГК и кетогексокиназе действовать как «система чувствительности к фруктозе», которая сигнализирует о том, что смешанная углеводная еда переваривается, и ускоряет утилизацию глюкозы. Однако фруктозо-6-фосфат (Ф6Ф) усиливает связывание ГК с помощью ГКРБ. Ф6Ф снижает фосфорилирование глюкозы ГК при гликогенолизе или глюконеогенезе .Ф1Ф и Ф6Ф связываются с одним и тем же сайтом на ГКРБ. Предполагается, что они продуцируют 2 разные конформации ГКРБ, одна способна связывать ГК, а другая нет.
Поджелудочной железы
Хотя большая часть глюкокиназы в организме находится в печени, меньшие количества в бета- и альфа-клетках поджелудочной железы, некоторых нейронах гипоталамуса и определённых клетках (энтероцитах) кишечника играют все более важную роль в регуляции углеводного обмена. В контексте функции глюкокиназы эти типы клеток вместе называются нейроэндокринными тканями, и у них есть общие аспекты регуляции и функции глюкокиназы, особенно общий нейроэндокринный промотор. Из нейроэндокринных клеток бета-клетки островков поджелудочной железы являются наиболее изученными и изученными. Вероятно, что многие из регуляторных отношений, обнаруженных в бета-клетках, будут также существовать в других нейроэндокринных тканях с глюкокиназой.
Сигнал для инсулина
В островковых бета-клетках активность глюкокиназы служит основным регулятором секреции инсулина в ответ на повышение уровня глюкозы в крови. По мере потребления Г6Ф увеличивающееся количество АТФ запускает серию процессов, которые приводят к высвобождению инсулина. Одним из непосредственных последствий учащенного клеточного дыхания является повышение концентраций НАДН и НАДФН (совместно именуемые НАД(Ф)Н). Этот сдвиг в окислительно-восстановительном статусе бета-клеток приводит к повышению уровня внутриклеточного кальция, закрытию каналов K-АТФ, деполяризации клеточной мембраны, слиянию секреторных гранул инсулина с мембраной и выбросу инсулина в кровь.
Именно в качестве сигнала для высвобождения инсулина глюкокиназа оказывает наибольшее влияние на уровень сахара в крови и общее направление метаболизма углеводов. Глюкоза, в свою очередь, влияет как на непосредственную активность, так и на количество глюкокиназы, продуцируемой бета-клетками.
Регуляция в бета-клетках
Глюкоза немедленно усиливает активность глюкокиназы за счет эффекта кооперативности.
Второй важный быстрый регулятор активности глюкокиназы в бета-клетках происходит за счет прямого белок-белкового взаимодействия между глюкокиназой и «бифункциональным ферментом» (фосфофруктокиназа-2/фруктоза-2,6-бисфосфатаза), который также играет роль в регуляции гликолиза. Эта физическая ассоциация стабилизирует глюкокиназу в каталитически благоприятной конформации (несколько противоположной эффекту связывания ГКРБ), что усиливает её активность.
Всего за 15 минут глюкоза может стимулировать транскрипцию GCK и синтез глюкокиназы посредством инсулина. Инсулин вырабатывается бета-клетками, но часть его действует на рецепторы инсулина B-типа бета-клеток, обеспечивая аутокринное усиление активности глюкокиназы с положительной обратной связью. Дальнейшая амплификация происходит под действием инсулина (через рецепторы А-типа) для стимуляции собственной транскрипции.
Транскрипция гена GCK инициируется через «вышестоящий», или нейроэндокринный, промотор. Этот промотор, в отличие от промотора печени, имеет элементы, гомологичные другим промоторам индуцированных инсулином генов. Среди возможных трансакционных факторов — Pdx-1 и PPARγ. Pdx-1 представляет собой фактор транскрипции гомеодомена, участвующий в дифференцировке поджелудочной железы. PPARγ — это ядерный рецептор, который реагирует на препараты глитазона повышением чувствительности к инсулину.
Связь с секреторными гранулами инсулина
Бо́льшая часть, но не вся глюкокиназа, обнаруженная в цитоплазме бета-клеток, связана с секреторными гранулами инсулина и митохондриями. «Связанная» доля быстро падает в ответ на повышение секреции глюкозы и инсулина. Было высказано предположение, что связывание служит цели, аналогичной цели печеночного регуляторного белка глюкокиназы, — защите глюкокиназы от деградации, так что она быстро становится доступной по мере повышения уровня глюкозы. Эффект заключается в усилении ответа глюкокиназы на глюкозу быстрее, чем это может сделать транскрипция[22].
Подавление глюкагона в альфа-клетках
Также было высказано предположение, что глюкокиназа играет роль в чувствительности к глюкозе альфа-клетками поджелудочной железы, но доказательства менее последовательны, и некоторые исследователи не нашли доказательств активности глюкокиназы в этих клетках. Альфа-клетки встречаются в островках поджелудочной железы, смешанные с бета-клетками и другими клетками. В то время как бета-клетки реагируют на повышение уровня глюкозы секрецией инсулина, альфа-клетки отвечают снижением секреции глюкагона. Когда концентрация глюкозы в крови падает до гипогликемического уровня, альфа-клетки выделяют глюкагон. Глюкагон — это белковый гормон, который блокирует действие инсулина на гепатоциты, вызывая гликогенолиз, глюконеогенез и снижая активность глюкокиназы в гепатоцитах. Степень, в которой подавление глюкозы глюкагона является прямым эффектом глюкозы через глюкокиназу в альфа-клетках или косвенным эффектом, опосредованным инсулином или другими сигналами от бета-клеток, все ещё не определена.
Гипоталамическая
В то время как все нейроны используют глюкозу в качестве топлива, некоторые чувствительные к глюкозе нейроны изменяют свою скорость возбуждения в ответ на повышение или понижение уровня глюкозы. Эти чувствительные к глюкозе нейроны сконцентрированы в основном в вентромедиальном ядре и дугообразном ядре гипоталамуса, которые регулируют многие аспекты гомеостаза глюкозы (особенно реакцию на гипогликемию), использование топлива, насыщение и аппетит, а также поддержание веса. Эти нейроны наиболее чувствительны к изменениям глюкозы в диапазоне 0,5-3,5 ммоль/л уровень глюкозы.
Глюкокиназа была обнаружена в головном мозге в основном в тех же областях, которые содержат нейроны, чувствительные к глюкозе, включая оба ядра гипоталамуса. Ингибирование глюкокиназы устраняет реакцию вентромедиального ядра на прием пищи. Однако уровень глюкозы в мозге ниже, чем в плазме, обычно 0,5-3,5. ммоль/л. Хотя этот диапазон соответствует чувствительности нейронов, чувствительных к глюкозе, он ниже оптимальной чувствительности к перегибу для глюкокиназы. Предположение, основанное на косвенных доказательствах, состоит в том, что нейрональная глюкокиназа каким-то образом подвергается воздействию уровня глюкозы в плазме даже в нейронах.
Энтероциты и инкретин
Хотя было показано, что глюкокиназа присутствует в определённых клетках (энтероцитах) тонкого кишечника и желудка, её функция и регуляция не изучены. Было высказано предположение, что и здесь глюкокиназа служит сенсором глюкозы, позволяя этим клеткам обеспечивать один из самых ранних метаболических ответов на поступающие углеводы. Предполагается, что эти клетки участвуют в функциях инкретина.
Клиническое значение
Поскольку инсулин является одним из, если не самым важным, регуляторов синтеза глюкокиназы, сахарный диабет всех типов снижает синтез и активность глюкокиназы по ряду механизмов. Активность глюкокиназы чувствительна к окислительному стрессу клеток, особенно бета-клеток.
Обнаружено около 200 мутаций гена глюкокиназы человека GCK, которые могут изменять эффективность связывания и фосфорилирования глюкозы, увеличивая или уменьшая чувствительность секреции инсулина бета-клетками в ответ на глюкозу и вызывая клинически значимую гипергликемию или гипогликемию.
Сахарный диабет
Мутации GCK снижают функциональную эффективность молекулы глюкокиназы. Гетерозиготность по аллелям со сниженной активностью ферментов приводит к более высокому порогу высвобождения инсулина и стойкой легкой гипергликемии. Это состояние называется диабетом 2 типа у молодых людей в зрелом возрасте (MODY2). В самом последнем обзоре мутации GCK, наблюдаемой у пациентов, говорится о 791 мутации, из которых 489, как полагают, вызывают диабет MODY и, следовательно, снижают функциональную эффективность молекулы глюкокиназы[23].
Гомозиготность по аллелям GCK с пониженной функцией может вызвать тяжелый врожденный дефицит инсулина, приводящий к стойкому неонатальному диабету.
Гиперинсулинемическая гипогликемия
Было обнаружено, что некоторые мутации усиливают секрецию инсулина. Гетерозиготность для увеличения функциональных мутаций снижает пороговое значение глюкозы, которое запускает высвобождение инсулина. Это создает гипогликемию различной формы, включая преходящий или стойкий врожденный гиперинсулинизм, или гипогликемию натощак или реактивную гипогликемию, возникающую в более старшем возрасте. В самом последнем обзоре мутаций GCK, которые наблюдались у пациентов, утверждалось, что 17 мутаций GCK вызывают гиперинсулинемическую гипогликемию[23].
Гомозиготность мутаций по усилению функции не обнаружена.
Исследовательская работа
Несколько фармацевтических компаний исследуют молекулы, активирующие глюкокиназу, в надежде, что она будет полезна при лечении диабета 1[24] и 2 типа[25][26][27].
Примечания
- “Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family”. Journal of Bioscience and Bioengineering. 99 (4): 320—30. April 2005. DOI:10.1263/jbb.99.320. PMID 16233797.
- “Molecular physiology of mammalian glucokinase”. Cellular and Molecular Life Sciences. 66 (1): 27—42. January 2009. DOI:10.1007/s00018-008-8322-9. PMID 18726182.
- Glucokinase and glycemic disease : from basics to novel therapeutics. — Basel: Karger, 2004. — 1 online resource (ix, 406 pages) с. — ISBN 1-4175-6491-1, 978-1-4175-6491-0, 978-3-318-01080-0, 3-318-01080-4.
- “Cloning and biochemical characterization of a novel mouse ADP-dependent glucokinase”. Biochemical and Biophysical Research Communications. 315 (3): 652—8. March 2004. DOI:10.1016/j.bbrc.2004.01.103. PMID 14975750.
- Glucokinase And Glycemic Disease: From Basics to Novel Therapeutics (Frontiers in Diabetes). — ISBN 3-8055-7744-3.
- Glucokinase // Encyclopedia of Molecular Medicine. — Hoboken : John Wiley & Sons, 2002. — ISBN 978-0-471-37494-7.
- “Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm”. Diabetes. 45 (2): 223—41. February 1996. DOI:10.2337/diabetes.45.2.223. PMID 8549869.
- “Glucose-induced conformational changes in glucokinase mediate allosteric regulation: transient kinetic analysis”. Biochemistry. 45 (24): 7553—62. June 2006. DOI:10.1021/bi060253q. PMID 16768451.
- “Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities”. Diabetes. 47 (3): 307—15. March 1998. DOI:10.2337/diabetes.47.3.307. PMID 9519733.
- “Identification of alkaline pH optimum of human glucokinase because of ATP-mediated bias correction in outcomes of enzyme assays”. Scientific Reports. 9 (1): 11422. August 2019. Bibcode:2019NatSR...911422S. DOI:10.1038/s41598-019-47883-1. PMID 31388064.
- Lunin VV, Li Y, Schrag JD, Iannuzzi P, Cygler M, Matte A (October 2004). “Crystal structures of Escherichia coli ATP-dependent glucokinase and its complex with glucose”. Journal of Bacteriology. 186 (20): 6915—27. DOI:10.1128/JB.186.20.6915-6927.2004. PMC 522197. PMID 15466045.
- “Structural model of human glucokinase in complex with glucose and ATP: implications for the mutants that cause hypo- and hyperglycemia”. Diabetes. 48 (9): 1698—705. September 1999. DOI:10.2337/diabetes.48.9.1698. PMID 10480597.
- “Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase”. Structure. 12 (3): 429—38. March 2004. DOI:10.1016/j.str.2004.02.005. PMID 15016359.
Beautiful structural pictures illustrating the conformational changes and potential regulatory mechanisms
- “A polymorphic (CA)n repeat element maps the human glucokinase gene (GCK) to chromosome 7p”. Genomics. 12 (2): 319—25. February 1992. DOI:10.1016/0888-7543(92)90380-B. PMID 1740341.
- “Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus”. Proceedings of the National Academy of Sciences of the United States of America. 89 (16): 7698—702. August 1992. Bibcode:1992PNAS...89.7698S. DOI:10.1073/pnas.89.16.7698. PMID 1502186.
- Glucokinase And Glycemic Disease: From Basics to Novel Therapeutics (Frontiers in Diabetes). — P. 18–30. — ISBN 3-8055-7744-3.
- “Differential expression and regulation of the glucokinase gene in liver and islets of Langerhans”. Proceedings of the National Academy of Sciences of the United States of America. 86 (20): 7838—42. October 1989. Bibcode:1989PNAS...86.7838I. DOI:10.1073/pnas.86.20.7838. PMID 2682629.
- “Transcriptional induction of glucokinase gene by insulin in cultured liver cells and its repression by the glucagon-cAMP system”. The Journal of Biological Chemistry. 264 (36): 21824—9. December 1989. DOI:10.1016/S0021-9258(20)88258-1. PMID 2557341.
- “Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut”. The Journal of Biological Chemistry. 269 (5): 3641—54. February 1994. DOI:10.1016/S0021-9258(17)41910-7. PMID 8106409.
- “Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy”. Human Mutation. 22 (5): 353—62. November 2003. DOI:10.1002/humu.10277. PMID 14517946.
- María Luz Cárdenas. Glucokinase : its regulation and role in liver metabolism. — New York: Springer-Verlag, 1995. — 210 pages с. — ISBN 1-57059-207-1, 978-1-57059-207-2, 3-540-59285-7, 978-3-540-59285-3.
- “Glucokinase is an integral component of the insulin granules in glucose-responsive insulin secretory cells and does not translocate during glucose stimulation”. Diabetes. 53 (9): 2346—52. September 2004. DOI:10.2337/diabetes.53.9.2346. PMID 15331544.
- “Evidence-based tailoring of bioinformatics approaches to optimize methods that predict the effects of nonsynonymous amino acid substitutions in glucokinase”. Scientific Reports. 7 (1): 9499. August 2017. DOI:10.1038/s41598-017-09810-0. PMID 28842611.
- TTP399 - VTV Therapeutics. VTV Therapeutics Corporate Website. Дата обращения: 8 апреля 2021.
- “Glucokinase activators in diabetes management”. Expert Opinion on Investigational Drugs. 17 (2): 145—67. February 2008. DOI:10.1517/13543784.17.2.145. PMID 18230050.
- “Assessing the potential of glucokinase activators in diabetes therapy”. Nature Reviews. Drug Discovery. 8 (5): 399—416. May 2009. DOI:10.1038/nrd2850. PMID 19373249.
- “A patent review of glucokinase activators and disruptors of the glucokinase--glucokinase regulatory protein interaction: 2011-2014”. Expert Opinion on Therapeutic Patents. 24 (8): 875—91. August 2014. DOI:10.1517/13543776.2014.918957. PMID 24821087.
Дальнейшее чтение
- Glaser, Benjamin. Familial Hyperinsulinism // GeneReviews. — Seattle WA : University of Washington, Seattle, 2013-01-24. — ISBN NBK1375.
- De León, Diva D. Permanent Neonatal Diabetes Mellitus // GeneReview / Diva D De León, Charles A Stanley. — Seattle WA : University of Washington, Seattle, 23 January 2014. — ISBN NBK1447.
