Биоортогональные реакции
Биоортогональные реакции — химические реакции, которые способны протекать внутри живых систем, не мешая естественным биохимическим процессам[1][2][3]. Функциональные группы, участвующие в биоортогональных реакциях, как правило, не встречаются в биомолекулах, быстро и селективно реагируют друг с другом в условиях живой клетки и при этом являются инертными по отношению к другим соединениям, которые присутствуют в организме. Термин был предложен Каролин Бертоцци в 2003 году[4]. Название реакций основано на переносном значении слова «ортогональный», то есть независимый от чего-либо, и обозначает взаимную независимость протекания искусственных и естественных процессов.
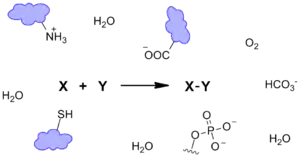
Несмотря на то, что в области органической химии открыто, изучено и описано большое множество химических реакций, практически ни одна из них не может быть проведена в клетке или организме так, чтобы затронуть лишь интересующее исследователя соединение (белок, ДНК, метаболит и др.). Так происходит потому, что биомолекулы содержат большое количество весьма похожих по реакционной способности функциональных групп (преимущественно нуклеофильных), и реакция, в которую вступает изучаемое соединение, неизбежно затронет и другие молекулы, содержащие подобные функциональные группы, что может не только не соответствовать задачам исследования, но и нарушать естественную работу клетки, делая невозможным её изучение[5]. В то же время проведение химических реакций внутри клетки является полезным инструментом исследования, поскольку позволяет помечать исследуемые биомолекулы флуоресцентными, аффинными и масс-спектрометрическими метками, которые в дальнейшем позволят наблюдать эти биомолекулы соответствующими методами исследования, например, при помощи флуоресцентного микроскопа. Биоортогональные реакции призваны заполнить этот пробел, поскольку они являются абсолютно чуждыми клетке, протекают между искусственно вводимыми функциональными группами и мало влияют на работу клетки[3].
Использование биоортогональной реакции на практике обычно осуществляют в две стадии. Сначала изучаемое соединение модифицируют биоортогональной функциональной группой внутри клетки. Затем в систему вводят низкомолекулярную метку, содержащую комплементарную функциональную группу, и в результате биоортогональной реакции происходит селективная модификация (мечение) данного соединения[3][5]. В дальнейшем введённая метка позволяет наблюдать за модифицированным субстратом.
В настоящее время при помощи биоортогональных реакций стало возможным изучение различных биомолекул, например, гликанов, белков[6] и липидов[7], в режиме реального времени в живых системах при отсутствии цитотоксичности. Было разработано несколько химических реакций, отвечающих требованиям биоортогональности, среди них 1,3-диполярное циклоприсоединение азидов к циклооктинам (называемое также безмедной клик-реакцией)[8], нитронов к циклооктинам[9], образование оксимов или гидразонов из карбонильных соединений[10], реакция тетразинов с циклооктенами[11], клик-реакция изонитрилов[12] и квадрициклановое лигирование[13].
История
Первое наблюдение селективной ковалентной модификации при помощи химической реакции в условиях клетки появилось в работе Р. Цяня и сотр., которые использовали флуоресцеин с двумя арсиновыми функциональными группами для селективной модификации белка, в который заранее вводили тетрацистеиновый фрагмент, практически отсутствующий в белках млекопитающих[14]. Данный подход позволил ввести в белок флуоресцентную метку небольшого размера, в то время как стандартным методом того времени было получение гибридов с зелёным флуоресцентным белком или его аналогами[15], обладающими значительно бо́льшим размером и, как следствие, порою мешающими нормальному функционированию исследуемого белка.
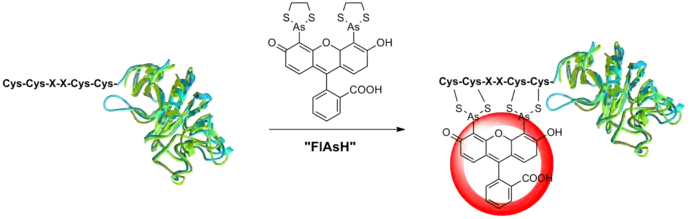
Данный подход побудил химиков искать химические реакции и функциональные группы, абсолютно чуждые природным соединениям, а биологов — изобретать пути введения биоортогональных функций в биомолекулы. Первым шагом стало осознание того, что биомолекулы содержат, в основном, нуклеофильные группы, в то время как электрофильные группы в них встречаются гораздо реже. Например, кетоны и альдегиды не встречаются в белках, в то же время они проявляют реакционную способность по отношению к гидразидным и оксиаминным группам, которые также не встречаются в биомолекулах. Благодаря этому, в конце 1990-х годов стала возможной модификация гликанов и белков через метаболически введённые кетон-содержащие сахара и аминокислоты внутри клетки. Кетоны и альдегиды, однако, присутствовали в низкомолекулярных метаболитах (сахара, пируват, пиридоксальфосфат и др.), то есть не были полностью биоортогональными[16].
В дальнейшем химики вели поиск биоортогональных реакций, протекающих между совершенно неприродными функциональными группами. Реакция Штаудингера стала первой химической реакцией, приспособленной для проведения модификаций внутри клетки. Азидная группа, вступающая в данную реакцию, относится к мягким электрофилам, которые не способны реагировать с жёсткими нуклеофилами, наиболее распространёнными в природе. Партнёром для азида стал фосфин — мягкий нуклеофил, предложенный Г. Штаудингером в 1919 году[17]. В 2000 году реакция Штаудингера была модифицирована К. Бертоцци и применена как биоортогональное лигирование по Штаудингеру для мечения широкого спектра биомолекул как в живых клетках, так и целых организмах[18].
В это же время стала развиваться клик-химия — химическая концепция, описывающая получение библиотек органических соединений при помощи быстрых и надёжных реакций, позволяющих собирать молекулы из небольших строительных блоков[19]. Среди нескольких реакций, удовлетворяющих данной концепции, азид-алкиновое циклоприсоединение, катализируемое медью, нашло широчайшее применение для модификации биомолекул in vitro[20]. Однако из-за токсичности медного катализатора такая реакция не могла быть использована в живых клетках или организмах. Группа К. Бертоцци создала некаталитический вариант данной реакции, известный как азид-алкиновое циклоприсоединение, облегчаемое напряжением (SPAAC), являющийся перспективной биоортогональной реакцией[8].
В настоящее время продолжается поиск новых биоортогональных реакций с целью получить возможность проведения параллельной модификации субстратов в одной и той же биологической системе.
Условия биоортогональности
В идеальном случае биоортогональная реакция должна соответствовать следующим частным условиям[3][4]:
- Селективность. Реакция должна протекать селективно между функциональными группами во избежание побочных процессов с участием биологических соединений.
- Биологическая инертность. Реагирующие группы и образующаяся связь не должны в какой-либо мере обладать реакционной способностью, способной нарушить естественные процессы, протекающие в изучаемом организме.
- Химическая инертность. Образующаяся ковалентная связь должна быть прочной и инертной по отношению к среде протекания реакции.
- Высокая скорость. Реакция должна быть быстрой, чтобы модификация наступила до расщепления или выведения метки из организма. Благодаря малому времени отклика быстрые реакции могут быть использованы для точного слежения за динамическими процессами в режиме реального времени.
- Биосовместимость. Реакция должна быть нетоксичной и протекать в биологических условиях, то есть при определенном рН, температуре и в водной среде.
- Доступная модификация субстрата. В идеальном случае биоортогональная функциональная группа должна вводиться в биомолекулу в ходе метаболизма или методами белковой инженерии. Функциональная группа должна быть небольшого размера и не нарушать нативное состояние биомолекулы.
Лигирование по Штаудингеру
Данная реакция была разработана группой К. Бертоцци в 2000 году на основе классической реакции Штаудингера между азидами и триарилфосфинами[18]. Эта реакция стала родоначальником области биоортогональной химии, поскольку реагирующие в ней группы (азиды, фосфины) не представлены в биомолекулах, однако теперь она не используется так же широко. Лигирование по Штаудингеру было использовано для модификации биомолекул как в живых клетках, так и в мышах[4].
Биоортогональность
В реакции Штаудингера азидная группа выступает как мягкий электрофил, реагируя с такими мягкими нуклеофилами, как фосфины. Большинство биологических нуклеофилов, напротив, являются жёсткими нуклеофилами, которые в реакцию с азидами не вступают. Кроме того, лигирование по Штаудингеру протекает в водной среде с образованием стабильного продукта[18].
Фосфины отсутствуют в природных биомолекулах[21] и не восстанавливают дисульфидные связи.
На примере лекарственных препаратов (азидотимидин) было показано, что азиды являются биосовместимыми. Маленький размер азидной группы позволяет легко вводить её в биомолекулы метаболическими путями[8].
Механизм
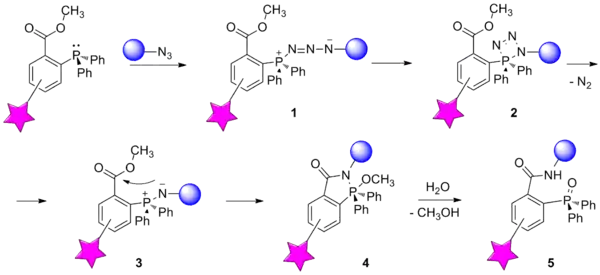
В основе реакции Штаудингера лежит нуклеофильная атака фосфина на концевой атом азота азидной группы с образованием фосфазида 1. Затем происходит превращение фосфазида 1 в иминофосфоран 3, сопровождающееся выделением азота, после чего наступает гидролиз иминофосфорана с образованием амина и фосфиноксида. Для применения в области биоконъюгации реакция была изменена путём введения сложноэфирной группы в орто-положение одного из арильных заместителей фосфина. В результате атаки образующегося иминофосфорана 3 на эту группу образуется бициклический продукт 4, гидролиз которого приводит к образованию устойчивой амидной связи между субстратом и вводимой меткой. Лимитирующей стадией реакции является атака молекулой фосфина азидной группы[22].
Ограничения
Основной недостаток данной реакции заключается в том, что фосфины медленно окисляются кислородом в живых системах. Кроме того, они, вероятно, метаболизируются цитохромами P450[4]. Лигирование по Штаудингеру протекает достаточно медленно, согласно кинетике второго порядка, с константой скорости около 0,0020 М−1 с−1[4]. Попытки ускорить нуклеофильную атаку введением электронодонорных групп в арильные заместители приводят к ускорению реакции, но также увеличивается и скорость окисления фосфинов.
Низкая скорость реакции требует увеличения концентрации используемого фосфина, из-за чего усиливается фоновый сигнал, производимый избыточным содержанием метки. Предпринимались усилия для преодоления этой проблемы: были синтезированы флуорогенные фосфины на основе флуоресцеина[23] и кумарина[24], действие которых основано на разгорании флуоресценции лишь после встраивания в биомолекулу, что позволяло получить больший скачок интенсивности излучения в результате протекания реакции. В данное время кинетика реакции остаётся серьёзным препятствием для её широкого использования.
Безмедное азид-алкиновое циклоприсоединение
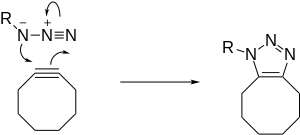
Безмедное азид-алкиновое циклоприсоединение — это биоортогональная реакция, разработанная К. Бертоцци как активированный вариант реакции Хьюсгена. В отличие от азид-алкинового циклоприсоединения, катализируемого медью (CuAAC, Cu-catalyzed azide-alkyne cycloaddition), данный вариант реакции протекает с более высокой скоростью за счёт снятия углового напряжения в молекуле циклооктина при образовании продукта присоединения. Поэтому эта реакция получила известность как азид-алкиновое циклоприсоединение, облегченное напряжением (SPAAC, Strain-promoted azide-alkyne cycloaddition). Данная модификация позволяет избежать использования токсичного медного катализатора и использовать безмедную реакцию в живых клетках и организмах.

Токсичность меди
Классическое азид-алкиновое циклоприсоединение, катализируемое медью, является очень быстрой и эффективной реакцией для биоконъюгации, однако оно не может быть использовано в живых клетках из-за токсичности ионов Cu+. Токсичность объясняется образованием активных форм кислорода, генерируемых медными катализаторами.
Проводилась оптимизация лигандов для предотвращения вредного воздействия на биомолекулы, однако было показано, что различное лигандное окружение в комплексах также влияет на метаболизм, так как вносит нежелательные изменения в клеточные процессы[25].
Биоортогональность
Азидная группа является биоортогональной, поскольку она достаточно мала (слабо влияет на проникновение молекулы в клетку), метаболически устойчива и не встречается в нативных биомолекулах. Хотя азиды не являются самыми реакционноспособными соединениями в 1,3-диполярном присоединении, их предпочитают использовать из-за отсутствия побочных реакций в условиях проведения модификации[26]. Циклооктиновый фрагмент имеет бо́льшие размеры, однако он обладает ортогональностью и стабильностью, необходимыми для модификации in vivo. Циклооктины являются наименьшими устойчивыми циклическими алкинами. Рассчитанное угловое напряжение в их циклах составляет 19,9 ккал/моль[27].
Механизм
Реакция протекает как стандартное 1,3-диполярное циклоприсоединение с согласованным перициклическим сдвигом электронов. Амбивалентная природа 1,3-диполя делает невозможным определение электрофильного и нуклеофильного центра в азиде, поэтому изображение направления перехода электронов не имеет смысла. Тем не менее, расчёты показывают, что внутренний атом азота несет самый большой отрицательный заряд[28].
Другие биоортогональные реакции
Диполярное циклоприсоединение нитронов
Безмедное азид-алкиновое циклоприсоединение было адаптировано для использования нитронов вместо азидов. При этом в качестве продуктов реакции образуются N-замещенные изоксазолины. Скорость реакции увеличивается в водной среде и подчиняется кинетике второго порядка с константой от 12 до 32 М−1 с−1 в зависимости от заместителей в нитроне. Несмотря на высокую скорость реакции, её недостатком являются трудности при введении нитрона в биомолекулу метаболическим мечением. Реакция применялась для модельной модификации пептида и пегилирования белка[9].
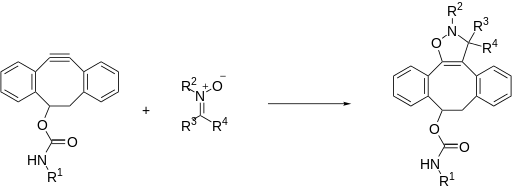
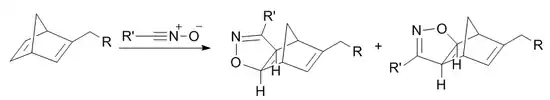
Циклоприсоединение норборнена
В 2009 году была разработана реакция диполярного циклоприсоединения нитрилоксидов к норборнену. В частности, она была применена для постсинтетической модификации олигонуклеотидов. Норборнен был выбран в качестве диполярофила благодаря балансу между промотируемой напряжением реакционной способности и устойчивостью. Недостатками данной реакции являются высокая электрофильность нитрилоксида, что приводит к побочным реакциям, а также низкая скорость реакции[29].
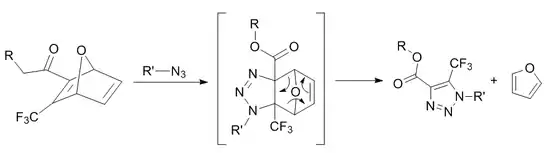
Циклоприсоединение оксанорборнадиена
Реакция циклоприсоединения оксанорборнадиена с азидами протекает с последующим элиминированием фурана по ретрореакции Дильса — Альдера. Напряжение цикла и электронная обеднённость оксанорборнадиена увеличивают его реакционную способность в лимитирующей стадии циклоприсоединения. Отщепление фурана происходит быстро с образованием устойчивого 1,2,3-триазола[30]. Предварительные исследования показали полезность данной реакции в модификации пептидов, а также она была использована при создании визуализирующих соединений в ОФЭКТ[31].
Реакция тетразинов с циклооктенами
Циклоприсоединение s-тетразинов и (E)-циклооктенов протекает как Реакция Дильса — Альдера, за которой следует ретрореакция Дильса — Альдера с выделением азота. Реакция протекает весьма быстро с константой скорости второго порядка, равной 2000 М−1 с−1 (в системе метанол — вода 9:1), что позволяет модифицировать биомолекулы в очень низких концентрациях.
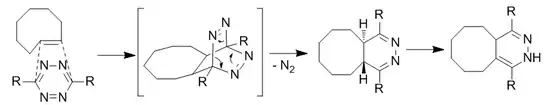
Проведенный расчёт показал, что энергия напряжения в (Z)-циклооктенах составляет 7,0 ккал/моль, что меньше, чем в циклооктане (12,4 ккал/моль), из-за потери двух трансаннулярных взаимодействий. Напротив, (E)-конфигурация двойной связи сильно увеличивает напряжение цикла (17,9 ккал/моль), что положительно сказывается на скорости реакции[32]. В качестве диенофила используется 3,6-диарил-s-тетразин, который содержит заместители для подавления взаимодействия с водой. Выделение азота на второй стадии делает реакцию необратимой[33].
Было обнаружено, что вода ускоряет реакцию тетразинов с циклооктенами. В качестве диенофилов использовали также норборнены, однако реакция протекала значительно медленнее (1 М−1 с−1 в водной среде). Реакцию тетразинов с (E)-циклооктеном применяли для мечения живых клеток флуоресцентной меткой[34] и сочетания полимеров[35].
[4+1]-Циклоприсоединение
Клик-реакция изонитрилов представляет собой [4+1]-циклоприсоединение, за которым следует ретрореакция Дильса-Альдера с выделением N2[12]. По данной причине реакция является необратимой. Продукт устойчив в том случае, если используется третичный изонитрил. В случае первичных и вторичных изонитрилов образуется имин, который затем быстро гидролизуется (на схеме изображено красным цветом).
Изонитрил является хорошей биоортогональной группой благодаря небольшому размеру, стабильности, нетоксичности и отсутствию в биологических системах. Однако, реакция [4+1]-циклоприсоединения протекает медленно с константой скорости второго порядка, равной 10−2 М−1 с−1.
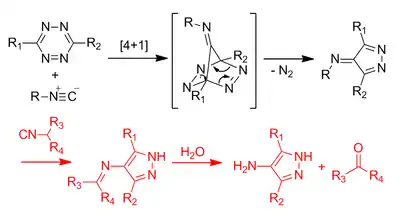
Фотоклик-реакция тетразола
Тетразол может подвергаться фотоиндуцированной реакции циклоэлиминирования с выделением азота. При этом образуется короткоживущий 1,3-диполь, вступающий в 1,3-диполярное циклоприсоединение с алкенами, приводя к пиразолиновым аддуктам[36].
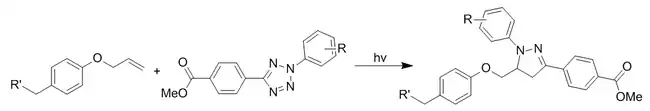
Фотоиндукция протекает при кратковременном облучении светом (длина волны зависит от тетразола). Время облучения подбирается так, чтобы снизить наносимый светом ущерб клеткам. Реакция ускоряется в водной среде и даёт единственный региоизомер. Достоинства такого подхода заключаются в возможности пространственного и временного контроля за реакцией. Использование реакции упрощается также тем, что алкены или тетразолы можно вводить в биомолекулы с использованием простых биологических методов. Кроме того, создан флуорогенный тетразол, позволяющий следить за степенью протекания реакции во времени[37].
Квадрициклановое лигирование
В ходе квадрицикланового лигирования напряженный квадрициклан вступает в [2+2+2]-циклоприсоединение с π-системами. Квадрициклан не встречается в нативных биомолекулах, не реагирует с ними (из-за насыщенности молекулы), обладает относительно малым размером и сильным напряжением (≈ 80 ккал/моль). Тем не менее, он очень стабилен при комнатной температуре и в водной среде при физиологическом рН. Он способен селективно реагировать с электронодефицитными π-системами, но не с простыми алкенами, алкинами или циклооктинами[13].
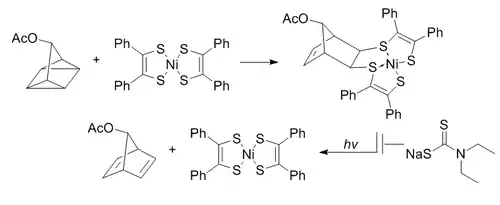
Бис(дитиобензил)никель(II) был выбран в качестве второго реагента в результате скрининга на реакционную способность. Для того, чтобы предотвратить фотоиндуцированную инверсию в норборнадиен, в реакцию добавляют диэтилдитиокарбамат, хелатирующий никель в образующемся продукте. Реакция протекает с константой скорости второго порядка, равной 0,25 М−1 с−1 (в водной среде).
С применением данной реакции, а также безмедного азид-алкинового циклоприсоединения и реакции образования оксимов, была создана методика одновременного мечения трёх субстратов без взаимной интерференции этих трёх реакций[13].
Применение
Некоторые биоортогональные реакции, в основном, лигирование по Штаудингеру и безмедное азид-алкиновое циклоприсоединение, широко используются в области биоконъюгации и химической биологии.
Мечение белков
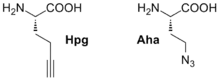
Клеточные системы синтеза белков, а также ферменты, могут внедрять в структуры белков неприродные аминокислоты, ошибочно принимая их за природные. В частности, было обнаружено, что замена метионина в питательной среде бактерий на гомопропаргилглицин (Hpg) или азидогомоаланин (Aha) позволила встроить эти синтетические аминокислоты в синтезируемые клеткой белки. Такие белки содержали биоортогональные функциональные группы — азид либо алкин, и введение в клетку биотина и флуоресцентных красителей, снабжённых комплементарной функциональной группой (фосфин, циклооктин либо азид) дало возможность селективно пометить и изучить данные белки. Кроме того, азидогомоаланин и гомопропаргилглицин были использованы одновременно для параллельной модификации двух различных типов белков[38].
Биоортогональная химия также нашла применение в профилировании белков по активности, которое позволяет изучать белки, имеющие сродство к определённому лиганду. Для этого лиганд помечается биоортогональной функциональной группой и вводится в клетку. После того, как связывание белков с лигандом произошло, клетка разрушается и совокупность всех белков вводится в реакцию с некоторой меткой, содержащей комплементарную реакционную группу. При этом метка вводится только в лиганд и позволяет отличить и выделить только те белки, которые связаны с этим лигандом[39].
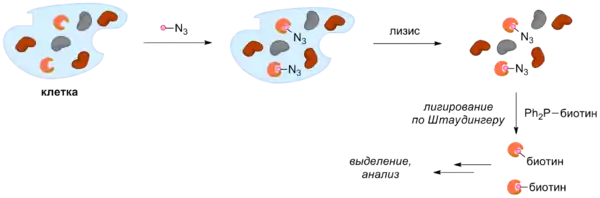
Мечение гликанов
Гликаны не закодированы генетически непосредственным образом и не обладают специфической активностью белков, поэтому к ним неприменимы методы генетического или аффинного введения меток. Однако, гликаны могут быть модифицированы метаболически при помощи модифицированных синтетических углеводов (сиаловой кислоты, N-ацетилгалактозамина, N-ацетилглюкозамина и др.), содержащих азидные группы. Гликаны со встроенными азидными группами вводили в лигирование по Штаудингеру с биотиновым реагентом и изучали методом проточной цитометрии[18].
Примечания
- Sletten E. M., Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality (англ.) // Angew. Chem. Int. Ed. — 2009. — Vol. 48, no. 38. — P. 6974–6998. — doi:10.1002/anie.200900942. — PMID 19714693.
- Prescher J. A., Dube D. H., Bertozzi C. R. Chemical remodelling of cell surfaces in living animals (англ.) // Nature. — 2004. — Vol. 430. — P. 873–877. — doi:10.1038/nature02791. — PMID 15318217.
- Prescher J. A., Bertozzi C. R. Chemistry in living systems (англ.) // Nature Chemical Biology. — 2005. — Vol. 1, no. 1. — P. 13–21. — doi:10.1038/nchembio0605-13. — PMID 16407987.
- Sletten E. M., Bertozzi C. R. From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions (англ.) // Acc. Chem. Res. — 2011. — Vol. 44, no. 9. — P. 666–676. — doi:10.1021/ar200148z. — PMID 21838330.
- Lim R. K. V., Qing Lin Q. Bioorthogonal chemistry: recent progress and future directions (англ.) // Chem. Commun. — 2010. — Vol. 46. — P. 1589–1600. — doi:10.1039/b925931g.
- Plass T., Milles S., Koehler C., Schultz C., Lemke E. A. Genetically Encoded Copper-Free Click Chemistry (англ.) // Angew. Chem. Int. Ed. — 2011. — Vol. 50, no. 17. — P. 3878–3881. — doi:10.1002/anie.201008178. — PMID 21433234.
- Neef A. B., Schultz C. Selective Fluorescence Labeling of Lipids in Living Cells (англ.) // Angew. Chem. Int. Ed. — 2009. — Vol. 48, no. 8. — P. 1498–1500. — doi:10.1002/anie.200805507. — PMID 19145623.
- Baskin J. M., Prescher J. A., Laughlin S. T., Agard N. J., Chang P. V., Miller I. A., Lo A., Codelli J. A., Bertozzi C. R. Copper-free click chemistry for dynamic in vivo imaging (англ.) // Proc. Natl. Acad. Sci. USA. — 2007. — Vol. 104, no. 43. — P. 16793–16797. — doi:10.1073/pnas.0707090104. — PMID 17942682.
- Ning X., Temming R. P., Dommerholt J., Guo J., Ania D. B., Debets M. F., Wolfert M. A., Boons G.-J., van Delft F. L. Protein Modification by Strain-Promoted Alkyne–Nitrone Cycloaddition (англ.) // Angew. Chem. Int. Ed. — 2010. — Vol. 49, no. 17. — P. 3065–3068. — doi:10.1002/anie.201000408. — PMID 20333639.
- Yarema K. J., Mahal L. K., Bruehl R. E., Rodriguez E. C., Bertozzi C. R. Metabolic Delivery of Ketone Groups to Sialic Acid Residues. Application to Cell Surface Glycoform Engineering (англ.) // J. Biol. Chem. — 1998. — Vol. 273, no. 47. — P. 31168–31179. — doi:10.1074/jbc.273.47.31168. — PMID 9813021.
- Blackman M. L., Royzen M., Fox J. M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity (англ.) // J. Am. Chem. Soc. — 2008. — Vol. 130, no. 41. — P. 13518–13519. — doi:10.1021/ja8053805. — PMID 18798613.
- Stöckmann H., Neves A. A., Stairs S., Brindle K. M., Leeper F. J. Exploring isonitrile-based click chemistry for ligation with biomolecules (англ.) // Org. Biomol. Chem. — 2011. — Vol. 9, no. 21. — P. 7303–7305. — doi:10.1039/C1OB06424J. — PMID 21915395.
- Sletten E. M., Bertozzi C. R. A Bioorthogonal Quadricyclane Ligation (англ.) // J. Am. Chem. Soc. — 2011. — Vol. 133, no. 44. — P. 17570–17573. — doi:10.1021/ja2072934. — PMID 21962173.
- Griffin B. A., Adams S. R., Tsien R. Y. Specific Covalent Labeling of Recombinant Protein Molecules Inside Live Cells (англ.) // Science. — 1998. — Vol. 281, no. 5374. — P. 269-272. — doi:10.1126/science.281.5374.269.
- Lippincott-Schwartz J., Patterson G. H. Development and Use of Fluorescent Protein Markers in Living Cells (англ.) // Science. — 2003. — Vol. 300, no. 5616. — P. 87-91. — doi:10.1126/science.1082520.
- Boyce M., Bertozzi C. R. Bringing chemistry to life (англ.) // Nature Methods. — 2011. — Vol. 8, iss. 8. — P. 638–642. — doi:10.1038/nmeth.1657.
- Staudinger H., Meyer J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. (нем.) // Helv. Chem. Acta. — 1919. — Bd. 2, H. 1. — S. 635–646. — doi:10.1002/hlca.19190020164.
- Saxon E., Bertozzi C. R. Cell Surface Engineering by a Modified Staudinger Reaction (англ.) // Science. — 2000. — Vol. 287, no. 5460. — P. 2007–2010. — doi:10.1126/science.287.5460.2007. — PMID 10720325.
- Kolb H. C., Finn M. G., Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions (англ.) // Angew. Chem. Int. Ed. — 2001. — Vol. 40, no. 11. — P. 2004–2021. — doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. — PMID 11433435.
- Meldal M., Tornøe C. W. Cu-Catalyzed Azide#Alkyne Cycloaddition (англ.) // Chem. Rev. — 2008. — Vol. 108, no. 8. — P. 2952-3015. — doi:10.1021/cr0783479.
- Chakraborty A., Wang D., Ebright Y. W., Ebright R. H. Azide-Specific Labelling of Biomolecules by Staudinger-Bertozzi Ligation: Phosphine Derivatives of Fluorescent Probes Suitable for Single-Molecule Fluorescence Spectroscopy (англ.) // Methods Enzymol. — 2010. — Vol. 472. — P. 19–30. — doi:10.1016/S0076-6879(10)72018-8.
- Lin F. L., Hoyt H. M., van Halbeek H., Bergman R. G., Bertozzi C. R. Mechanistic Investigation of the Staudinger Ligation (англ.) // J. Am. Chem. Soc. — 2005. — Vol. 127, no. 8. — P. 2686–2695. — doi:10.1021/ja044461m. — PMID 15725026.
- Hangauer M. J., Bertozzi C. R. A FRET-Based Fluorogenic Phosphine for Live-Cell Imaging with the Staudinger Ligation (англ.) // Angew. Chem. Int. Ed. — 2008. — Vol. 47, no. 13. — P. 2394–2397. — doi:10.1002/anie.200704847.
- Lemieux G. A., de Graffenried C. L., Bertozzi C. R. A Fluorogenic Dye Activated by the Staudinger Ligation (англ.) // J. Am. Chem. Soc. — 2003. — Vol. 125, no. 16. — P. 4708–4709. — doi:10.1021/ja029013y. — PMID 12696879.
- Kennedy D. C., McKay C. S., Legault M. C. B., Danielson D. C., Blake J. A., Pegoraro A. F., Stolow A., Mester Z., Pezacki J. P. Cellular Consequences of Copper Complexes Used To Catalyze Bioorthogonal Click Reactions (ут) // J. Am. Chem. Soc. — 2011. — Т. 133, № 44. — С. 17993–18001. — doi:10.1021/ja2083027. — PMID 21970470.
- Huisgen R. 1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates (англ.) // J. Org. Chem. — 1976. — Vol. 41, no. 3. — P. 403–419. — doi:10.1021/jo00865a001.
- Schoenebeck F., Ess D. H., Jones G. O., Houk K. N. Reactivity and Regioselectivity in 1,3-Dipolar Cycloadditions of Azides to Strained Alkynes and Alkenes: A Computational Study (англ.) // J. Am. Chem. Soc. — 2009. — Vol. 131, no. 23. — P. 8121–8133. — doi:10.1021/ja9003624. — PMID 19459632.
- Gold B., Shevchenko N. E., Bonus N., Dudley G. B., Alabugin I. V. Selective Transition State Stabilization via Hyperconjugative and Conjugative Assistance: Stereoelectronic Concept for Copper-Free Click Chemistry (англ.) // J. Org. Chem. — 2012. — Vol. 77, no. 1. — P. 75–89. — doi:10.1021/jo201434w. — PMID 22077877.
- Gutsmiedl K., Wirges C. T., Ehmke V., Carell T. Copper-Free “Click” Modification of DNA via Nitrile Oxide−Norbornene 1,3-Dipolar Cycloaddition (англ.) // Org. Lett. — 2009. — Vol. 11, no. 11. — P. 2405–2408. — doi:10.1021/ol9005322. — PMID 19405510.
- van Berkel S. S., Dirks A. J., Debets M. F., van Delft F. L., Cornelissen J. J. L. M., Nolte R. J. M., Rutjes F. P. J. T. Metal-Free Triazole Formation as a Tool for Bioconjugation (англ.) // ChemBioChem. — 2007. — Vol. 8, no. 13. — P. 1504–1508. — doi:10.1002/cbic.200700278. — PMID 17631666.
- van Berkel S. S., Dirks A. J., Meeuwissen S. A., Pingen D. L. L., Boerman O. C., Laverman P., van Delft F. L., Cornelissen J. J. L. M., Rutjes F. P. J. T. Application of Metal-Free Triazole Formation in the Synthesis of Cyclic RGD–DTPA Conjugates (англ.) // ChemBioChem. — 2008. — Vol. 9, no. 11. — P. 1805–1815. — doi:10.1002/cbic.200800074. — PMID 18623291.
- Bach R. D. Ring Strain Energy in the Cyclooctyl System. The Effect of Strain Energy on [3 + 2] Cycloaddition Reactions with Azides (англ.) // J. Am. Chem. Soc. — 2009. — Vol. 131, no. 14. — P. 5233–5243. — doi:10.1021/ja8094137. — PMID 19301865.
- Blackman M. L., Royzen M., Fox J. M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity (англ.) // J. Am. Chem. Soc. — 2008. — Vol. 130, no. 41. — P. 13518–13519. — doi:10.1021/ja8053805.
- Devaraj N. K., Ralph Weissleder R., Hilderbrand S. A. Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging (англ.) // Bioconjugate Chem. — 2008. — Vol. 19, no. 12. — P. 2297–2299. — doi:10.1021/bc8004446. — PMID 19053305.
- Hansell C. F., Espeel P., Stamenović M. M., Barker I. A., Dove A. P., Du Prez F. E., O’Reilly R. K. Additive-Free Clicking for Polymer Functionalization and Coupling by Tetrazine–Norbornene Chemistry (англ.) // J. Am. Chem. Soc. — 2011. — Vol. 133, no. 35. — P. 13828–13831. — doi:10.1021/ja203957h. — PMID 21819063.
- Lim R. K. V., Lin Q. Photoinducible Bioorthogonal Chemistry: A Spatiotemporally Controllable Tool to Visualize and Perturb Proteins in Live Cells (англ.) // Acc. Chem. Res. — 2011. — Vol. 44, no. 9. — P. 828–839. — doi:10.1021/ar200021p. — PMID 21609129.
- Song W., Wang Y., Qu J.,Lin Q. Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via “Photoclick Chemistry” in Bacterial Cells (англ.) // J. Am. Chem. Soc. — 2008. — Vol. 130, no. 30. — P. 9654–9655. — doi:10.1021/ja803598e. — PMID 18593155.
- Beatty K. E., Tirrell D. A. Two-color labeling of temporally defined protein populations in mammalian cells (англ.) // Bioorg. Med. Chem. Lett. — 2008. — Vol. 18, no. 22. — P. 5995–5999. — doi:10.1016/j.bmcl.2008.08.046. — PMID 18774715.
- Hang H. C., Loureiro J., Spooner E., van der Velden A. W. M., Kim Y.-M., Pollington A. M., Maehr R., Starnbach M. N., Ploegh H. L. Mechanism-Based Probe for the Analysis of Cathepsin Cysteine Proteases in Living Cells (англ.) // ACS Chem. Biol. — 2006. — Vol. 1, no. 11. — P. 713–723. — doi:10.1021/cb600431a.
Литература
- Prescher J. A., Bertozzi C. R. Chemistry in living systems // Nature Chemical Biology. — 2005. — Т. 1, № 1. — С. 13—21. — doi:10.1038/nchembio0605-13. — PMID 16407987.
- Sletten E. M., Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality // Angew. Chem. Int. Ed. — 2009. — Т. 48, № 38. — С. 6974—6998. — doi:10.1002/anie.200900942.
- Lim R. K. V., Lin Q. Bioorthogonal chemistry: recent progress and future directions // Chem. Commun. — 2010. — Т. 46, № 10. — С. 1589—1600. — doi:10.1039/b925931g. — PMID 20177591.
- Boyce M., Bertozzi C. R. Bringing chemistry to life // Nature Methods. — 2011. — Т. 8, № 8. — С. 638–642. — doi:10.1038/nmeth.1657.
