Азид-алкиновое циклоприсоединение
Азид-алкиновое циклоприсоединение — реакция между азидами и алкинами с образованием 1,2,3-триазолов.
Реакция впервые была описана Михаэлем в 1893 г., который обнаружил, что при нагреве эфирного раствора фенилазида и диметилового эфира ацетилендикарбоновой кислоты в запаянной ампуле (8 ч при 100 °C) образуется замещенный триазол[1]. Некаталитический вариант реакции был исследован Хьюсгеном в начале 1960-х в рамках изучения реакций 1,3-диполярного присоединения[2][3]. В литературе он получил название «реакция Хьюсгена».
В классическом варианте реакция идет по механизму 1,3-диполярного присоединения ведёт к образованию смеси изомерных 1,4- и 1,5-дизамещенных 1,2,3-триазолов:

Широкое развитие реакция получила после открытия катализа медью(I) в лабораториях Мельдаля[4] и Шарплесса[5] в 2002 году, став важнейшей реакцией в рамках концепции клик-химии[6]. Улучшенный вариант, ускоряемый напряжением циклооктинового фрагмента, является перспективным направлением исследования данной реакции. Благодаря открытым модификациям, реакция вошла в число клик-реакций.
Реакция, катализируемая медью (CuAAC)
О катализе одновалентной медью было впервые сообщено в независимых публикациях Мортена Мельдаля[4] и Барри Шарплесса[5]. Каталитический вариант реакции не протекает синхронно, а имеет постадийный механизм, поэтому не может называться реакцией Хьюсгена, хотя в литературе иногда встречается такое название. Благодаря введению катализатора реакция получила несколько преимуществ, позволивших применить её в различных биотехнологических приложениях, и стала известна под аббревиатурой CuAAC (Cu-catalyzed azide-alkyne cycloaddition).
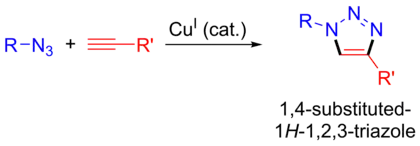
Преимущества
- Каталитический вариант реакции протекает приблизительно в 107 раз быстрее по сравнению с некаталическим вариантом, что позволяет проводить реакцию азид-алкинового циклоприсоединения при температурах, близких к комнатной.
- На протекание реакции слабо влияют заместители при азидной и алкиновой группах.
- Для реакции подходит широкий спектр растворителей, в том числе вода, а также водно-органические смеси.
- Медь-каталитический вариант является региоселективным и даёт 1,4-дизамещенные 1,2,3-триазолы в качестве единственных продуктов.[7]
Механизм
Постадийный механизм медь-катализируемой реакции протекает через промежуточное образование ацетиленидов меди. По этой причине высокую реакционную способность в данной реакции демонстрируют лишь терминальные алкины. Одновременно атом меди оказывает активирующее влияние на азид путём его координации, что также определяет региоселективность реакции. Далее происходит образование шестичленного металлацикла, который претерпевает восстановительное элиминирование с образованием триазолил-медного производного. В результате гидролиза последнего образуется 1,4-дизамещенный 1,2,3-триазол.[8]
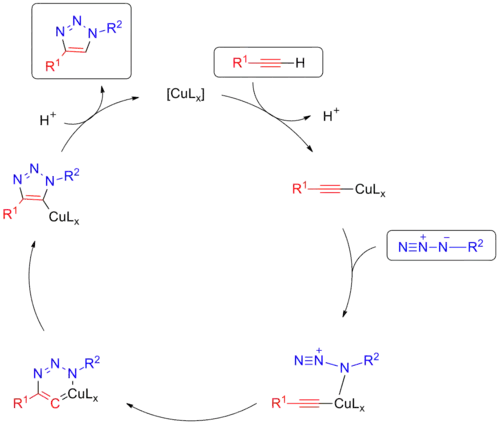
Каталитические системы
Реакция протекает в присутствии разнообразных источников Cu(I). При условии, что реагенты находятся в растворе или даже агрегированной смеси и что одновалентная медь не исчезла в результате диспропорционирования или окисления кислородом воздуха, продукты обычно получаются с высоким выходом. Для постоянного поддержания достаточной концентрации Cu(I) используют соединения Cu(II) в присутствии избытка восстановителя, который не только генерирует Cu(I), но и делает реакцию менее восприимчивой к кислороду.
Для реакций, протекающих в водной среде, наиболее часто используют систему CuSO4 — аскорбат натрия. Другим источником одновалентной меди являются её соли (CuBr, CuI). При этом в качестве среды выступают органические растворители (тетрагидрофуран, пиридин, ДМСО, ацетонитрил и др.). Для повышения растворимости этих солей применяют комплексы типа [Cu(CH3CN)4]PF6, (EtO)3P•CuI. Если медь используется непосредственно в одновалентном состоянии, необходимо предпринимать меры для изолирования реакции от кислорода воздуха, например, проведением реакции в инертной атмосфере или с добавлением восстановителя.
Реже каталитические количества одновалентной меди вводят по реакции компропорционирования Cu(0) и Cu(II), при этом источником нульвалентной меди служат медные проволоки, порошки, наночастицы и проч.[9]
Для ускорения реакции и стабилизации каталитической частицы применяют триазольные (TBTA)[10], а также некоторые другие лиганды[9].
Реакция, промотируемая напряжением (SPAAC)
Ускорение реакции может быть достигнуто не только путём использования катализатора, но и повышением реакционной способности алкина. Данный подход применен для создания азид-алкинового циклоприсоединения, промотируемого напряжением цикла (strain-promoted azide-alkyne cycloaddition, SPAAC)[11]. Введение в реакцию с азидами напряженного циклооктина улучшает кинетику реакции и позволяет проводить циклоприсоединение в отсутствие цитотоксичного медного катализатора.

Механизм
Реакция протекает как стандартное 1,3-диполярное циклоприсоединение с асинхронным согласованным перициклическим сдвигом электронов. Амбивалентная природа 1,3-диполя делает невозможным определение электрофильного и нуклеофильного центра в азиде, поэтому изображение направления перехода электронов не имеет смысла. Тем не менее, расчёты показывают, что внутренний атом азота несет самый большой отрицательный заряд.[12]
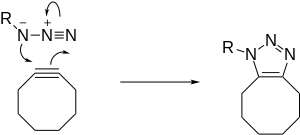
Региоселективность
Несмотря на то, что в результате реакции образуется смесь двух региоизомерных триазолов, это не является существенным недостатком для большинства современных приложений. Высокой региоселективности можно добиться при использовании медь-катализируемой реакции с терминальными алкинами.
Разработка циклооктинов
| Циклооктин | Константа скорости второго порядка (М−1с−1) |
|---|---|
| OCT | 0,0024 |
| ALO | 0,0013 |
| MOFO | 0,0043 |
| DIFO | 0,076 |
| DIBO | 0,057 |
| BARAC | 0,96 |
| DIBAC (ADIBO) | 0,31 |
| DIMAC | 0,0030 |
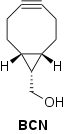
| BCN | 0,14-0,29 |

OCT был первым циклооктином, разработанным для безмедного азид-алкинового циклоприсоединения.[13] В то время как алкины линейного строения нереакционноспособны при физиологических температурах, OCT легко реагировал с алкинам в биологических условиях, не оказывая при этом токсического действия. Однако, он имел низкую растворимость в воде, а кинетика едва превосходила кинетику лигирования по Штаудингеру. Как улучшенный вариант был разработан ALO (aryl-less octyne, «октин без арила»), но он также реагировал медленно.[14]
Монофторированный (MOFO)[14] и дифторированный (DIFO)[15] циклооктины создавались для увеличения скорости реакции путём введения электроноакцепторных атомов фтора в пропаргильное положение. Фтор — удобная акцепторная группа как с точки зрения синтетической доступности, так и биологической инертности. В частности, он не может образовывать акцептор Михаэля, дающий побочные реакции с биологическими нуклеофилами.
DIBO (дибензоциклооктин) получил два конденсированных бензольных кольца, что привело к повышению углового напряжения циклооктинового фрагмента. Было предположено, что сопряжение арильных фрагментов с тройной связью увеличит реакционную способность соединения.
Добавление ещё одной двойной связи в циклооктин приводило к неустойчивым соединениям, поэтому группа Бертоцци предложила циклооктин BARAC (биарилазациклооктинон) с амидной связью, обладающей частичной двоесвязностью за счёт резонанса. Кроме того, добавление гетероатома в молекулу увеличивает растворимость и улучшает фармакокинетику молекулы. BARAC реагирует с азидами достаточно быстро, поэтому отмывание избытка реагента не требуется, что критично в тех приложениях, где такие отмывки невозможны (слежение за динамическими процессами в реальном времени, мечение биомолекул в организмах). Хотя BARAC исключительно полезен, по причине нестабильности его нужно хранить при 0°С в темном месте, в отсутствие кислорода.[16]
Дальнейшние изменения структуры BARAC приводят к DIBAC (ADIBO) с меньшими стерическими затруднениями у алкиновой функции.[17] Соединение, комбинирующее в себе наличие сопряженного бензольного кольца и двух атомов фтора в пропаргильном положении, (DIFBO, дифторбензоциклооктин) оказалось неустойчивым.[18]
Проблемы с использованием DIFO в исследованиях in vivo на мышах могут иллюстрировать трудности в создании биоортогональных реакций. Хотя DIFO был очень реакционноспособным в модификации клеток, в мышах он показал себя с худшей стороны из-за связывания с сывороточным альбумином. Гидрофобность циклооктина является причиной его взаимодействия с клеточными мембранами и белками сыворотки, что сильно понижает его доступные концентрации. В качестве водорастворимого аналога с повышенной полярностью и улучшенной фармакокинетикой был предложен DIMAC (диметоксиазациклооктин).
Проверялись также другие методы создания дополнительного напряжения циклооктинового цикла. В частности, хорошие результаты показал BCN (бициклононин), в котором данный эффект достигался за счет введения в молекулу конденсированного трёхчленного цикла.[19]
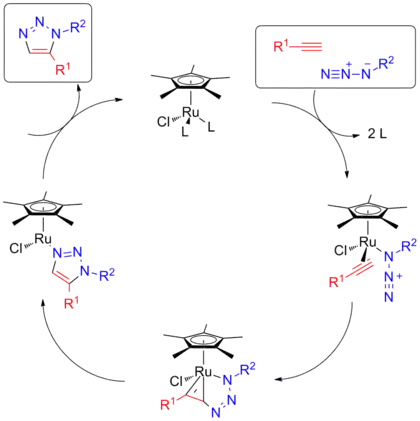
Реакция, катализируемая рутением (RuAAC)
Реакция азид-алкинового циклоприсоединения, катализируемая комплексами рутения, (RuAAC) приводит к образованию 1,5-дизамещенных триазолов[20]. Важным отличием от CuAAC является возможность синтеза полностью замещенных триазолов, поскольку дизамещенные алкины также могут участвовать в данной реакции. В качестве катализаторов обычно используют Cp*RuCl(PPh3)2, Cp*Ru(COD)and Cp*[RuCl4]. Применяются также катализаторы, содержащие циклопентадиенильный лиганд (Cp), однако лучшие результаты получены с участием пентаметилциклопентадиенильного лиганда (Cp*).
Механизм
Предложенный механизм включает образование активных каталитических частиц [Cp*RuCl], после чего происходит обмен лигандов на азид и алкин, окислительное присоединение с образованием рутенацикла и восстановительное элиминирование с формированием триазольного продукта. В данном процессе атом азота формирует связь с более доступным атомом углерода алкина, что определяет региоселективность реакции[21].
Реакция, катализируемая серебром (AgAAC)
Реакция азид-алкинового циклоприсоединения может катализироваться P,O-комплексами серебра(I) с преимущественным образованием 1,4-дизамещённых триазолов при комнатной температуре. Соли серебра(I) не катализируют данную реакцию.[22][23]
Примечания
- Michael A. Ueber die Einwirkung von Diazobenzolimid auf Acetylendicarbonsauremethylester (нем.) // J. Prakt. Chem. — 1893. — Bd. 48. — S. 94–95.
- Huisgen R. 1,3-Dipolar Cycloadditions. Past and Future (англ.) // Angew. Chem. Int. Ed. — 1963. — Vol. 2, no. 10. — P. 565–598. — doi:10.1002/anie.196305651.
- Huisgen R. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions (англ.) // Angew. Chem. Int. Ed. — 1963. — Vol. 2, no. 11. — P. 633–645. — doi:10.1002/anie.196306331.
- Tornøe C. W., Christensen C., Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides (англ.) // J. Org. Chem. — 2002. — Vol. 67, no. 9. — P. 3057–3064. — doi:10.1021/jo011148j. — PMID 11975567.
- Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes (англ.) // Angew. Chem. Int. Ed. — 2002. — Vol. 41, no. 14. — P. 2596–2599. — doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. — PMID 12203546.
- Kolb H. C., Finn M. G., Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions (англ.) // Angew. Chem. Int. Ed. — 2001. — Vol. 40, no. 11. — P. 2004–2021. — doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. — PMID 11433435.
- Brase S., Banert K. Organic Azides: Syntheses and Applications. — Wiley, 2009. — С. 270. — 536 с. — ISBN 978-0-470-51998-1.
- Wu P., Fokin V. V. Catalytic Azide–Alkyne Cycloaddition: Reactivity and Applications (англ.) // Aldrichimica Acta. — 2007. — Vol. 40, no. 1. — P. 7–17.
- Meldal M., Tornøe C. W. Cu-Catalyzed Azide#Alkyne Cycloaddition (англ.) // Chem. Rev. — 2008. — Vol. 108, no. 8. — P. 2952-3015. — doi:10.1021/cr0783479.
- Chan T. R., Hilgraf R., Sharpless K. B., Fokin V. V. Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis (англ.) // Org. Lett. — 2004. — Vol. 6, no. 17. — P. 2853–2855. — doi:10.1021/ol0493094.
- Baskin J. M., Prescher J. A., Laughlin S. T., Agard N. J., Chang P. V., Miller I. A., Lo A., Codelli J. A., Bertozzi C. R. Copper-free click chemistry for dynamic in vivo imaging (англ.) // Proc. Natl. Acad. Sci. USA. — 2007. — Vol. 104, no. 43. — P. 16793–16797. — doi:10.1073/pnas.0707090104. — PMID 17942682.
- Gold B., Shevchenko N. E., Bonus N., Dudley G. B., Alabugin I. V. Selective Transition State Stabilization via Hyperconjugative and Conjugative Assistance: Stereoelectronic Concept for Copper-Free Click Chemistry (англ.) // J. Org. Chem. — 2012. — Vol. 77, no. 1. — P. 75–89. — doi:10.1021/jo201434w. — PMID 22077877.
- Agard N. J., Prescher J. A., Bertozzi C. R. A Strain-Promoted [3 + 2] Azide-Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems (англ.) // J. Am. Chem. Soc. — 2004. — Vol. 126, no. 46. — P. 15046–15047. — doi:10.1021/ja044996f. — PMID 15547999.
- Agard N. J., Baskin J. M., Prescher J. A., Lo A., Bertozzi C. R. A Comparative Study of Bioorthogonal Reactions with Azides (англ.) // ACS Chem. Biol. — 2006. — Vol. 1, no. 10. — P. 644–648. — doi:10.1021/cb6003228. — PMID 17175580.
- Codelli J. A., Baskin J. M., Agard N. J., Bertozzi C. R. Second-Generation Difluorinated Cyclooctynes for Copper-Free Click Chemistry (англ.) // J. Am. Chem. Soc. — 2008. — Vol. 130, no. 34. — P. 11486–11493. — doi:10.1021/ja803086r. — PMID 18680289.
- Jewett J. C., Sletten E. M., Bertozzi C. R. Rapid Cu-Free Click Chemistry with Readily Synthesized Biarylazacyclooctynones (англ.) // J. Am. Chem. Soc. — 2010. — Vol. 132, no. 11. — P. 3688–3690. — doi:10.1021/ja100014q. — PMID 20187640.
- Kuzmin A., Poloukhtine A., Wolfert M. A., Popik V. V. Surface Functionalization Using Catalyst-Free Azide-Alkyne Cycloaddition (англ.) // Bioconjugate Chem. — 2010. — Vol. 21, no. 11. — P. 2076–2085. — doi:10.1021/bc100306u. — PMID 20964340.
- Sletten E. M., Nakamura H., Jewett J. C., Bertozzi C. R. Difluorobenzocyclooctyne: Synthesis, Reactivity, and Stabilization by β-Cyclodextrin (англ.) // J. Am. Chem. Soc. — 2010. — Vol. 132, no. 33. — P. 11799–11805. — doi:10.1021/ja105005t. — PMID 20666466.
- Dommerholt J., Schmidt S., Temming R., Hendriks L. J. A., Rutjes F. P. J. T., van Hest J. C. M., Lefeber D. J., Friedl P., van Delft F. L. Readily Accessible Bicyclononynes for Bioorthogonal Labeling and Three-Dimensional Imaging of Living Cells (англ.) // Angew. Chem. Int. Ed. — 2010. — Vol. 49, no. 49. — P. 9422–9425. — doi:10.1002/anie.201003761. — PMID 20857472.
- Zhang L., Chen X., Xue P., Sun H. H. Y., Williams I. D., Sharpless K. B., Fokin V. V., Jia G. Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides (англ.) // J. Am. Chem. Soc. — 2005. — Vol. 127, no. 46. — P. 15998–15999. — doi:10.1021/ja054114s. — PMID 16287266.
- Boren B. C., Narayan S., Rasmussen L. K., Zhang L., Zhao H., Lin Z., Jia G., Fokin V. V. Ruthenium-Catalyzed Azide-Alkyne Cycloaddition: Scope and Mechanism (англ.) // J. Am. Chem. Soc. — 2008. — Vol. 130, no. 44. — P. 8923–8930. — doi:10.1021/ja0749993. — PMID 18570425.
- McNulty J., Keskar K., Vemula R. The First Well-Defined Silver(I)-Complex-Catalyzed Cycloaddition of Azides onto Terminal Alkynes at Room Temperature (англ.) // Chem. Eur. J. — 2011. — Vol. 17, iss. 52. — P. 14727–14730. — doi:10.1002/chem.201103244.
- McNulty J., Keskar K. Discovery of a Robust and Efficient Homogeneous Silver(I) Catalyst for the Cycloaddition of Azides onto Terminal Alkynes (англ.) // Eur. J. Org. Chem. — 2012. — Vol. 2012, iss. 28. — P. 5462–5470. — doi:10.1002/ejoc.201200930.