Миастения
Миастения Гравис (лат. myasthenia gravis; др.-греч. μῦς «мышца» + ἀσθένεια «бессилие, слабость») — аутоиммунное нервно-мышечное заболевание, характеризующееся патологически быстрой утомляемостью поперечнополосатых мышц.
| Миастения Гравис | |
|---|---|
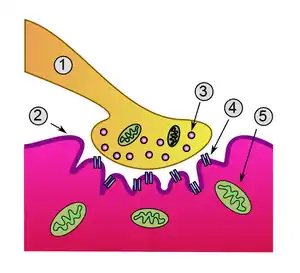 Схема нервно-мышечного синапса: 1. Пресинаптическая терминаль 2. Сарколемма 3. Синаптический пузырёк 4. Никотиновый ацетилхолиновый рецептор 5. Митохондрия | |
| МКБ-11 | 8C60 |
| МКБ-10 | G70.0 |
| МКБ-10-КМ | G70.0 и G70.00 |
| МКБ-9 | 358.0 |
| МКБ-9-КМ | 358.00[1][2] и 358.0[1][2] |
| OMIM | 254200 |
| DiseasesDB | 8460 |
| MedlinePlus | 000712 |
| eMedicine |
neuro/232 emerg/325 (emergency), med/3260 (pregnancy), oph/263 (eye) |
| MeSH | D009157 |
Миастения Гравис (астенический бульбарный паралич, астеническая офтальмоплегия, ложный бульбарный паралич, болезнь Эрба — Гольдфлама) является классическим аутоиммунным заболеванием человека. Основным клиническим проявлением миастении является синдром патологической мышечной утомляемости (усиление проявлений миастении после физической нагрузки и уменьшение их после отдыха).
История
Впервые заболевание было описано Томасом Уиллисом в 1672 году. Антитела при миастении впервые были выделены Strauss в 1960 году. Окончательно аутоиммунное происхождение болезни было доказано Patrick и Lindstrom в опыте на кроликах, иммунизированных Torpedo californica AChR, у которых появились признаки миастении.
Эпидемиология
Заболевание обычно начинается в возрасте 20—40 лет; чаще болеют женщины. В последнее время заболеваемость миастенией растёт, на сегодняшний момент распространённость составляет приблизительно 5—10 человек на 100 000 населения.
Этиология
Миастения бывает как врождённой, так и приобретённой. Причиной врождённой миастении являются мутации в генах различных белков, отвечающих за построение и работу нервно-мышечных синапсов. В синапсах (в частности, в концевых пластинках нервно-мышечных синапсов) ацетилхолинэстераза присутствует в виде тетрамера изоформы T, присоединённого к коллагеноподобному белку, который кодируется отдельным геном COLQ[3]. Мутация этого гена является одной из наиболее распространённых причин наследственной миастении (myasthenia gravis)[4]. Другой распространённой причиной миастении являются различные мутации субъединиц никотинового рецептора ацетилхолина[5].
Иногда, чаще у молодых, возникает опухоль вилочковой железы, которую удаляют хирургическим путём.
Патогенез
В механизме развития миастении играют роль аутоиммунные процессы, обнаружены антитела в мышечной ткани и вилочковой железе. Часто поражаются мышцы век, появляется птоз, который варьирует по степени выраженности в течение дня; поражаются жевательные мышцы, нарушается глотание, изменяется походка. Больным вредно нервничать, так как это вызывает боль в груди и одышку.
Провоцирующим фактором может являться стресс, перенесённая ОРВИ, а также нарушение функции иммунной системы организма, которое ведёт к образованию антител против собственных клеток организма — ацетилхолиновых рецепторов постсинаптической мембраны нервно-мышечных соединений (синапсов). По наследству аутоиммунная миастения не передаётся.
Чаще всего заболевание проявляется во время переходного возраста у девочек (11-13 лет), реже встречается у мальчиков в этом же возрасте. Все чаще выявляется заболевание у детей дошкольного возраста (5-7 лет).
Симптоматика

Существует несколько форм миастении (глазная, генерализованная, бульбарная и миастенический синдром Ламберта — Итона (при раке лёгкого и др.). Заболевание чаще начинается с глазных симптомов (опущение век, двоение). Особенностью является динамичность симптомов: утром птоз может быть меньше, чем вечером, двоение меняется по выраженности. Затем чаще присоединяется слабость проксимальных отделов мышц конечностей (трудно подняться по лестнице, подняться со стула, поднимать руки вверх). При этом на фоне физической нагрузки слабость отчётливо нарастает во всех группах мышц (после пробы с 10 приседаниями слабость увеличивается не только в мышцах ног, но и рук, усиливается птоз). Могут присоединяться бульбарные нарушения (на фоне длительного разговора или во время приёма пищи голос приобретает гнусавый оттенок, появляется дизартрия, трудно выговаривать «Р», «Ш», «С». После отдыха эти явления проходят). Далее бульбарные нарушения могут стать более выраженными (появляется нарушение глотания, поперхивания, попадание жидкой пищи в нос).
Приём антихолинэстеразных препаратов (калимин, прозерин) в значительной мере улучшает состояние больных — такие больные стараются принимать пищу на пике действия антихолинэстеразных препаратов.
Диагностика
В стандартных случаях диагностика миастении включает:
- Клинический осмотр и выяснение истории болезни.
- Функциональная проба на выявление синдрома патологической мышечной утомляемости. Электромиографическое исследование: декремент-тест
- Прозериновая проба
- Повторный декремент-тест для выявления реакции на прозерин
- Клинический осмотр для выявления обратимости миастенических изменений на фоне прозерина
- Анализ крови на антитела к ацетилхолиновым рецепторам и антитела к титину
- Компьютерная томография органов переднего средостения (вилочковой железы, синоним: тимуса).
В случаях сложной дифференциальной диагностики проводятся игольчатая электромиография, исследование проводящей функции нервов, электромиография отдельных мышечных волокон (джиттер), биохимические исследования (креатинфосфокиназа, лактат, пируват, 3-гидроксибутират).
Лечение
В случаях лёгкой впервые выявленной миастении и глазной формы в лечении применяется только калимин и препараты калия.
Калимин 60Н по 1 таб 3 раза в день с интервалом не менее 6 часов. Хлористый калий по 1 г 3 раза в день или калий-нормин по 1 таб 3 раза в день.
В случаях выраженной мышечной слабости или наличии бульбарных нарушений применяется глюкокортикоидная терапия: преднизолон в дозе 1 мг/кг веса строго через день в утренние часы (обычные дозы составляют 60-80 мг в сутки, минимально эффективные дозы составляют 50 мг в сутки через день).
Одна таблетка преднизолона содержит 5 мг, соответственно суточная доза преднизолона составляет 12—16 таблеток. Таблетка Метипреда (одно из коммерческих названий метилпреднизолона) содержит 4 мг, но по эффективности она равна 1 таб преднизолона 5 мг, поэтому в пересчёте на метипред количество таблеток составляет те же 12—16 таб, а суммарная доза будет меньше.
Преднизолон 60 мг утром через день.
Приём преднизолона длительный, ремиссия может наступить через 1—2 месяца, далее доза преднизолона по 0,5 таб снижается до поддерживающей дозы 10—40 мг через день. А затем медленно с осторожностью по 0,25 таблетки до полного исключения препарата.
Приём преднизолона требует контроля сахара крови и контроля со стороны участкового терапевта (артериальное давление, профилактика стероидной язвы, остеопороза).
В первые 1-2 года от начала заболевания при генерализованной форме миастении проводится оперативное вмешательство по удалению вилочковой железы (тимэктомия). Эффект от тимэктомии развивается в интервале 1-12 месяцев с момента тимэктомии, оценка эффективности тимэктомии производится через 1 год.
В пожилом возрасте, при недостаточной эффективности терапии преднизолоном, при невозможности назначения преднизолона и при отмене преднизолона назначается цитостатическая терапия. В лёгких случаях — азатиоприн по 50 мг (1 таб) 3 раза в день. В более серьезных случаях — циклоспорин (сандиммун) по 200—300 мг в сутки или селлсепт по 1000—2000 мг в сутки.
При обострении миастении допустимо и оправдано проведение плазмафереза и введение внутривенного иммуноглобулина. Плазмаферез целесообразно проводить по 500 мл через день N5-7 с замещением плазмой или альбумином.
Иммуноглобулин вводится внутривенно в дозе 5—10 г в сутки до суммарной дозы 10—30 г, в среднем 20 г. Вводится иммуноглобулин медленно, по 15 капель в минуту.
В 2017 году регуляторы систем здравоохранения США (FDA), ЕС (EMA) и Японии (MHLW) одобрили лекарство Soliris (экулизумаб) в качестве лечения для пациентов с генерализованной миастенией (гМГ).
В декабре 2019 года голландская «Ардженекс» (Argenx) предложила «Вивгарт» (Vyvgart, эфгартигимод[6]) — новый препарат, предназначенный для лечения взрослых пациентов с генерализованной миастенией гравис и аутоантителами к ацетилхолиновому рецептору (AChR). Эфгартигимод (efgartigimod), будучи блокатором неонатального Fc-рецептора (FcRn), способствует снижению рециркуляции патогенных иммуноглобулинов G (IgG) и увеличению их деградации[7].
Противопоказания при миастении
- Чрезмерные физические нагрузки
- Инсоляции (ограничить пребывание под прямыми солнечными лучами)
- Препараты магния (магнезия, панангин, аспаркам)
- Курареподобные миорелаксанты
- Нейролептики и транквилизаторы (кроме грандаксина), ГОМК
- Мочегонные (кроме верошпирона и других спиронолактонов)
- Антибиотики:
- аминогликозиды (гентамицин, стрептомицин, неомицин, канамицин, мономицин, тобрамицин, сизомицин, амикацин, дидезоксиканамицин-В, нетилмицин)
- фторхинолоны (эноксацин, норфлоксацин, ципрофлоксацин, офлоксацин, флероксацин, ломефлоксацин, спарфлоксацин), а тетрациклин — под наблюдением врача
- Фторсодержащие кортикостероиды (дексаметазон, дексазон, полькортолон)
- Производные хинина
- D-пеницилламин.
Прогноз
Ранее миастения была тяжёлым заболеванием с высокой летальностью — 30—40 %. Однако при современных методах диагностики и лечения летальность стала минимальной — менее 1 %, около 80 % на фоне правильного лечения достигают полного выздоровления или же ремиссии. Заболевание является хроническим, требует тщательного наблюдения и лечения.
Примечания
- база данных Disease ontology (англ.) — 2016.
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- OMIM Entry - * 603033 - COLLAGENIC TAIL OF ENDPLATE ACETYLCHOLINESTERASE; COLQ
- Myasthenic Syndromes
- Myasthenic Syndromes
- VYVGART- efgartigimod alfa injection (англ.). DailyMed. U. S. National Library of Medicine.
- Фон Ройсс, Татьяна. «Вивгарт»: новый препарат для лечения миастении гравис. Эфгартигимод разработки Argenx — сильный конкурент «Солириса». Mosmedpreparaty.ru. «Мосмедпрепараты» (19 декабря 2021).
Литература
- Ильина H. А., Кузин М. И., Сачков В. И., Сигаев В. В. Миастения // Большая медицинская энциклопедия : в 30 т. / гл. ред. Б. В. Петровский. — 3-е изд. — М. : Советская энциклопедия, 1981. — Т. 15 : Меланома — Мудров. — 576 с. : ил.
