Транскриптомика одиночных клеток
Транскрипто́мика одино́чных кле́ток (англ. single-cell transcriptomics) — область биологических исследований, в которой основным инструментом служат методы количественного анализа экспрессии генов в индивидуальных клетках. Изучение транскриптома отдельных клеток позволяет решить проблему «усреднённых» данных, которые получаются при анализе тотальной РНК, выделенной из образца[1]. Секвенирование РНК одиночных клеток сделало возможными анализ клеточного многообразия в популяциях клеток, считавшихся ранее однородными, например, были получены новые данные в областях иммунологических, эмбриологических и онкологических исследований[2][3][4]. Развитие технологий с 2009 года, когда впервые было произведено секвенирование транскриптома одиночных клеток, по наше время позволило увеличить производительность эксперимента от единиц до сотен тысяч клеток, что существенно повысило точность получаемых данных[5].
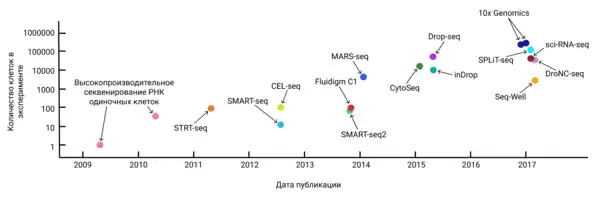
* 2009 — Впервые проведено секвенирование РНК отдельных клеток.
* 2010 — Количество клеток в эксперименте увеличено до ~100 за счёт параллельной обработки образцов.
* 2011 — Технология STRT-Seq: улучшенное покрытие 5' конца[6].
* 2012 — Технология SMART-Seq: амплификация с полным покрытием[7], технология CEL-seq — избавление от систематических ошибок при ПЦР с помощью in vitro транскрипции.
* 2013 — Технологии SMART-Seq2 — удешевление процесса, улучшенная чувствительность. Секвенирование на коммерческой платформе Fluidigm C1 с использованием микрочастиц жидкости позволило увеличить количество клеток в эксперименте до ~1000,[7].
* 2014 — Появление технологий с использованием уникальных молекулярных идентификаторов (англ. UMI): MARS-Seq, CEL-Seq UMI, STRT-Seq UMI[8].
* 2015 — Технологии CytoSeq, Drop-seq, inDrop: увеличение производительности до сотен клеток за счёт использования наночастиц жидкости и иммобилизованных праймеров[9].
* 2017 — усовершенствование технологий, in situ баркодирование. Производительность увеличена до ~1000000 клеток.
Технология анализа транскриптома единичных клеток с помощью количественной ПЦР
Количественная ПЦР применяется для анализа транскриптома единичных клеток реже, чем секвенирование. Для проведения эксперимента требуется выделение одиночных клеток, их лизис, обратная транскрипция РНК. Этот метод достаточно чувствительный, он может быть использован на микрофлюидных технологиях, однако он не позволяет исследовать весь транскриптом, а только детектировать количество конкретных транскриптов, к которым подобраны зонды или праймеры. При этом уровень экспрессии изучаемых генов определяется не абсолютно, а относительно референсного гена[10][11].
Количественная ПЦР может также применяться для валидации данных секвенирования РНК[12][13].
Технология секвенирования РНК отдельных клеток
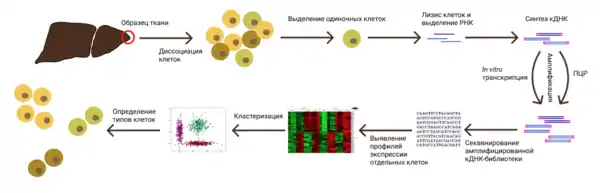
Общий алгоритм секвенирования РНК одиночных клеток включает в себя 8 последовательных этапов[9]:
- Выделение единичных клеток;
- Лизис единичных клеток;
- Выделение интересующей фракции РНК с помощью специальных праймеров;
- Синтез кДНК с помощью обратной транскрипции;
- Амплификация кДНК;
- Подготовка библиотеки кДНК;
- Секвенирование;
- Биоинформатическая обработка полученных прочтений и анализ данных.
Различные методы подготовки библиотек для секвенирования РНК одиночных клеток отличаются по своей специфичности, точности, стоимости и другим параметрам. Например, Smart-seq2 отличается высокой чувствительностью, а Drop-seq и другие микрофлюидные технологии с использованием микрочастиц очень высокопроизводительны[14][9].
Выделение отдельных клеток
Перед разделением клеток нужно прервать контакты между ними и избавиться от межклеточного вещества. Это может быть достигнуто с помощью ферментирования образца ткани, а также путём специфических манипуляций, таких как, например, лазерная захватывающая микродиссекция[6], которая позволяет выделять клетки из образца твёрдой ткани с помощью лазера. После получения клеточной суспензии клетки разделяют с помощью различных методов[7].
- Метод предельного разбавления (англ. limiting dilution). Этот метод заключается в том, что из разбавленной клеточной суспензии пипеткой выделяются отдельные клетки. Процедура производится вручную[7].
- Микроманипуляция (англ. micromanipulation). Это метод ручного выделения клеток из ткани под микроскопом. Он обычно используется для выделения клеток ранних эмбрионов или отбора клеток некультивируемых микроорганизмов[7].
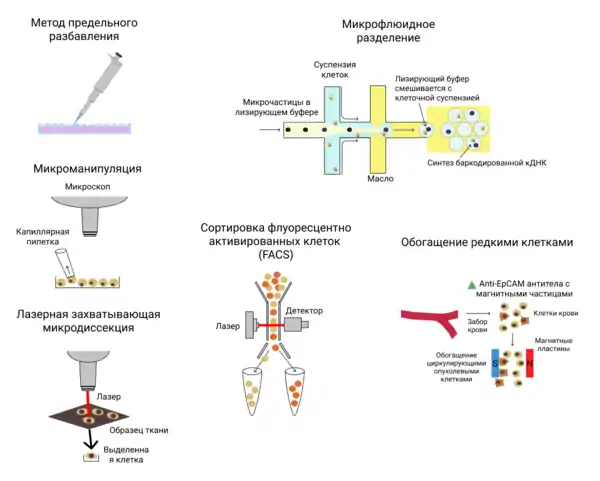
- Сортировка флуоресцентно активированных клеток (англ. fluorescence-activated cell sorting (FACS)). Это часто используемый высокопроизводительный метод. Перед сортировкой клетки метят с помощью моноклональных антител к поверхностным маркерам, антитела снабжены флуоресцентными метками. Механическая сортировка происходит следующим образом. Суспензию клеток помещают в прибор, который выбрасывает капли, содержащие по одной клетке, через наконечник специального «распылителя». Эти капли проходят через луч лазера, и, в зависимости от типа флуоресцентной метки (или их совокупности) на клетке, капля получает заряд, который заставляет её притягиваться к одной из отклоняющих пластин, в результате чего она попадает в конкретный собирающий сосуд. Возможно также производить негативный отбор и выделять клетки, не несущие маркеров. Данный метод не позволяет работать с образцами малого объёма (требуется подавать на вход около 10000 клеток) и требует наличия антител к поверхностным белкам[7].
- Микрофлюидные технологии[15] (англ. microfluidic technology). Это также высокопроизводительный метод, использующийся наряду с FACS. Он основан на изоляции клеток в микрокаплях буфера, погружённых в гидрофобную среду (масло). Разделение происходит в трубках, диаметр которых подобран так, чтобы капли, содержащие клетки, не сливались друг с другом. Капля раствора, в которую помещается клетка, содержит реакционную смесь для лизиса, выделения РНК и синтеза кДНК. Такой принцип используется в коммерческом аппарате Fluidigm C1 и позволяет обрабатывать до 1000 клеток параллельно[7].
- Микрофлюидные технологии с использованием микрочастиц (англ. microdroplet-based microfluidics). Данный принцип также основан на изоляции клеток в гидрофильных микрочастицах жидкости в гидрофобной среде, но помимо клеток, в каплю попадает также твёрдая микрочастица с иммобилизированными на ней праймерами. Так же как и в предыдущем случае, в каплях содержатся изолированные клетки, лизирующий буфер и реакционная смесь для обратной транскрипции, в том числе изолированные на микрочастицах праймеры. Этот метод используется в коммерческом аппарате Chromium system from 10× Genomics. Он позволяет работать с каплями меньшего объёма, чем микрофлюидные технологии без микрочастиц, и обрабатывать от 1000 до 1000000 клеток[7].
- Микрофлюидные технологии с использованием микроячеек. Данный метод основан на распределении клеток по ячейкам такого размера, который допускает попадание в них не более одной клетки и микрочастицы с иммобилизованными праймерами[15].
- Выделение редких циркулирующих опухолевых клеток. Некоторые специфические задачи требуют выделения из популяции очень малочисленных клеток. Например, выделение из крови циркулирующих опухолевых клеток (англ. circulating tumor cells, CTCs) производится с помощью добавления к образцу крови антител с магнитными частицами и выделения меченых клеток с помощью магнита. Для опухолевых клеток эпителиального происхождения используются антитела к CD45− и EpCAM+[7].
Лизис клеток и выделение РНК
Клетки обычно лизируют химически, помещая их в лизирующий буфер. Лизирующие буферы могут различаться по качеству сохранения содержимого клетки и эффективности дальнейших процедур, проводимых с лизатом[16]. Оптимальные протоколы лизиса одиночных клеток эукариот и прокариот также различны, так как требуется разрушить массивную и часто покрытую защитными оболочками клеточную стенку прокариот, при этом не повредив выделяемый материал[17].
Выделение РНК при подготовке образцов происходит не отдельным техническим этапом, а получается за счёт использования специальных праймеров для инициации обратной транскрипции[1].
Получение кДНК
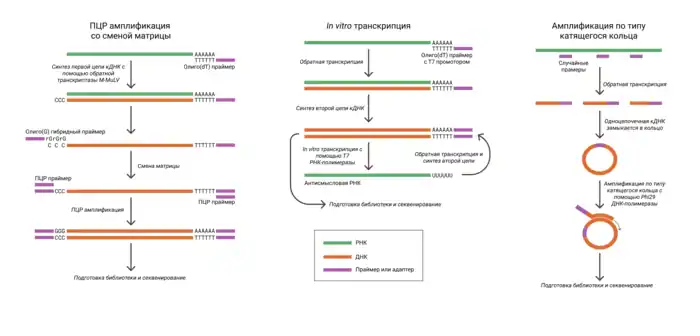
После выделения РНК необходимо получить из неё комплементарную ДНК (кДНК) с помощью обратной транскрипции[6]. Первая цепь кДНК синтезируется при помощи специально спроектированной версии обратной транскриптазы вируса лейкемии мышей M-MuLV[18]. Для инициации синтеза используются праймеры, имеющие в своей последовательности баркоды, иногда уникальные молекулярные идентификаторы и последовательности, позволяющие отобрать интересующую нас фракцию РНК. Обычно нужно избавиться от рРНК и тРНК, которые составляют до 95 % выделенной тотальной РНК клетки. Этого можно достичь, используя праймеры с поли(dT)-участком, что позволяет выделить полиаденилированную фракцию. Однако при этом теряется неполиаденилированная РНК (длинные некодирующие РНК и другие), поэтому в ряде протоколов, например, SUPeR-seq, в последовательности праймеров после поли(dT)-участка добавляется несколько (5—6) случайных нуклеотидов.
Синтез второй цепи кДНК осуществляется различными способами. Часто используется метод смены матрицы (англ. template switching), например, в технологиях STRT, Smart-seq и Smart-seq2. Он основан на свойстве ревертазы M-MuLV добавлять на 3’-конец синтезируемой цепи нематричные остатки цитозина. Соответственно, это делает возможным синтез второй цепи с поли(dG)-праймеров[18].
Баркоды и уникальные молекулярные идентификаторы
Технология высокопроизводительного секвенирования предполагает совместное секвенирование библиотек, полученных из разных клеток. Поэтому для различения транскриптов, пришедших из каждой конкретной клетки, используются уникальные клеточные баркоды[7][9] . В экспериментах по дифференциальной экспрессии помимо баркодов используются так называемые уникальные молекулярные идентификаторы (англ. unique molecular identifiers, UMIs). UMI представляет собой последовательность из 4—8 случайных нуклеотидов (например, 5 нуклеотидов дают 45=1024 уникальные последовательности). Сочетание UMI и клеточного баркода статистически получается уникальным для каждого транскрипта, что позволяет сравнивать уровни экспрессии генов по количеству UMI, «пришитых» к транскриптам определённого типа. Баркоды и уникальные молекулярные идентификаторы вносятся в образец на этапе обратной транскрипции, так как составляют часть праймера для синтеза первой цепи кДНК[7].
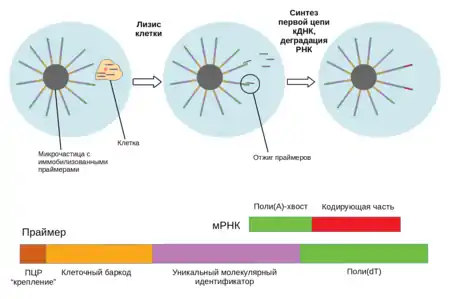
Амплификация кДНК
В ряд технологий, таких как MARS-seq, CEL-seq и CEL-seq2, для амплификации кДНК используется in vitro транскрипция (англ. in vitro transcription, IVT)[6]. Это способ основан на транскрипции кДНК фаговой полимеразой Т7 и повторении этапа обратной транскрипции. Для осуществления in vitro транскрипции в поли(dT)-праймер вносится промотор Т7. Увеличение количества кДНК в данном случае происходит линейно[6].
Амплификация кДНК может также осуществляться с помощью полимеразной цепной реакции (ПЦР), например, в Drop-seq, SCRB-seq, SMART-seq и SMART-seq2. Однако этот метод часто вносит искажения в отношение количества транскриптов. С этими искажениями позволяет бороться использование уникальных молекулярных идентификаторов[7].
Для работы с прокариотическими клетками используются также специальные методы, такие как амплификация по типу катящегося кольца[17].
Подготовка геномной библиотеки и секвенирование
В зависимости от способа подготовки библиотеки происходит секвенирование полноразмерных транскриптов, или фракции, обогащённой 3’- или 5’-фрагментами[6][7]. Обогащение полноразмерными транскриптами (технологии SMART-seq, SMART-seq2) требуется при изучении альтернативного сплайсинга и однонуклеотидных полиморфизмов, тогда как секвенирование 3’-фрагментов (технологии CEL- seq, CEL-seq2, MARS-seq) и 5’-фрагментов (технология STRT) подходят для выявления дифференциальной экспрессии. Эти методы, как правило, используют уникальные молекулярные идентификаторы. Подготовленные библиотеки обрабатывают методами секвенирования нового поколения (англ. next generation sequencing, NGS), часто используется секвенирование на платформе Illumina. Полученные «сырые» прочтения обрабатывают методами биоинформатики[7].
Анализ данных
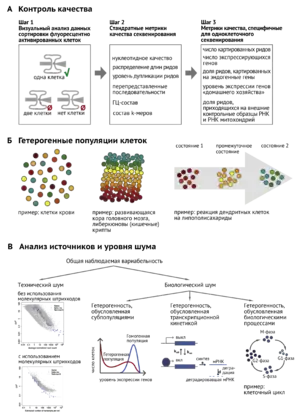
А. Контроль качества результатов эксперимента
Б. Примеры исследования гетерогенных популяций клеток: разные клеточные типы, спектр состояний клеток в ткани, спектр состояний клеток при развитии или ином биологическом процессе
В. Анализ источников и уровня шума: биологического и технического
Первоочередной задачей при биоинформатическом анализе результатов секвенирования РНК одиночных клеток является получение матрицы экспрессий генов из прочтений секвенатора. После получения такой матрицы имеют место несколько направлений анализа[7]:
- Анализ на клеточном уровне: кластеризация, классификация и определение клеточных траекторий;
- Анализ на уровне генов: определение дифференциально экспрессирующихся генов, регуляторных сетей.
Получение матрицы экспрессии генов
Стандартный протокол обработки прочтений, получаемых при секвенировании, включает в себя несколько шагов (в скобках приведены программы, использующиеся на каждом этапе)[19]:
- Контроль качества прочтений (FastQC, Kraken);
- Картирование прочтений на референсный геном (TopHat2[20], HISAT[21] и другие);
- Подсчёт количества транскриптов исследуемых генов для каждой из клеток (используя характеристики покрытия FPKM/TPM или число уникальных молекулярных идентификаторов);
- При необходимости (например, если два набора данных были получены в разных местах и разными учёными) — поправка систематической ошибки (kBET[22]);
- В случае, если использовался протокол без уникальных молекулярных идентификаторов, необходима нормализация матрицы (SCnorm[23], SAMstrt[24]);
- Восстановление пропущенных данных (импутация) (MAGIC[25], Autoimpute[26]).
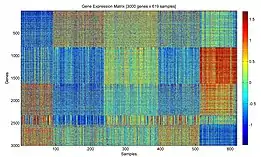

Контроль качества прочтений
При картировании обеспечивается контроль качества прочтения транскриптома каждой клетки, клетки с низким качеством прочтения исключаются из дальнейшего анализа[27]. Для контроля качества могут использоваться разные метрики:
- количество прочтений на клетку;
- количество найденных генов;
- отношение количества всех прочтений к количеству прочтений митохондриальной РНК (высокое отношение может означать утечку цитоплазматических РНК или протекание апоптоза в клетке);
- калибровка количества всех прочтений к РНК, количество и последовательность которой известна RNA spike-in;
- использование UMI (уникальных молекулярных идентификаторов).
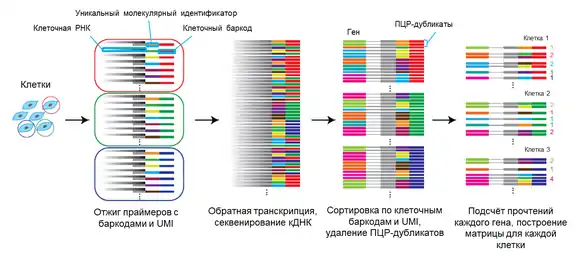
Обработка данных с применением UMI и клеточных баркодов
Последовательно выполняются следующие шаги[28]:
- Проводится обратная транскрипция, UMI и клеточные баркоды находятся на праймере и входят в состав кДНК;
- Прочтения сортируются по UMI и клеточным баркодам, удаляются ПЦР-дубликаты: прочтения с одинаковым клеточным баркодом и UMI;
- Для каждой клетки строится матрица, характеризующая количество прочтений (у каждого оставшегося прочтения уникальное сочетание «клеточный баркод + UMI») каждого найденного гена.
Кластеризация
С целью выявления клеточных субпопуляций обычно проводится кластеризация клеток по схожести их профилей экспрессии генов[29]. Эта кластеризация может проводиться многими способами: методом k-средних[30], с использованием графа ближайших соседей[31], иерархической кластеризацией[32] и некоторыми другими. Несмотря на обилие подходов, кластеризация получается не всегда: структура данных может скрываться за техническим шумом или систематическими ошибками[33][34]; также анализ затрудняется из-за проклятия размерности. Для сглаживания этих эффектов размерность транскриптомного пространства, элементами которого являются клетки, понижается[29].
Уменьшение размерности
При выполнении формальных математических операций классификации, поиска корреляций принимается, что каждая клетка — это вектор в n-мерном пространстве, где n соответствует числу анализируемых генов, а координаты клетки — это уровни экспрессий соответствующих генов в ней[35]. Как уже было сказано, снижение размерности может помочь восстановить структуру данных и уменьшить шумы, и потому размерность векторов экспрессий имеет смысл понижать (при помощи метода главных компонент[36], t-SNE[37], многомерного шкалирования[38], UMAP[39] и других).
Дифференциальная экспрессия генов
Важной задачей является поиск дифференциально экспрессирующихся генов, то есть таких генов, которые статистически достоверно экспрессируются в разных группах клеток с разной силой. Такие гены часто характеризуют особенности рассматриваемых клеток и являются их маркерами[19]. Сначала для идентификации дифференциальной экспрессии использовали инструменты, созданные для работы с транскриптомикой тканей и органов; сейчас существует ряд методов (MAST[40], SCDE[41]), созданных для поиска дифференциальной экспрессии в данных секвенирования именно отдельных клеток.
Генные регуляторные сети
Генная регуляторная сеть — это совокупность молекулярных регуляторов, взаимодействующих друг с другом и другими веществами в клетке, регулируя уровни экспрессии[42]. Эти регуляторы играют центральную роль в морфогенезе частей тела и органов живых организмов и являются одним из центральных предметов изучения эволюционной биологии развития. Генную регуляторную сеть можно представить как граф, в котором вершины — это гены, а рёбра — это их ко-регуляция. Существуют методы, определяющие регуляторные сети при помощи поиска корреляций между экспрессиями генов, однако такой подход не позволяет детектировать нелинейные взаимодействия, поэтому сейчас возникли подходы, основанные на машинном обучении[43], вероятностных моделях[44], а также теории информации[45].
Поиск траекторий дифференцировок клеток
Клетки постоянно находятся в динамических процессах и реагируют на различные воздействия окружающей среды. Эти процессы сопровождаются и изменением профиля транскрипции клетки. Сама постановка эксперимента по секвенированию РНК одиночных клеток позволяет захватывать клетки в их разные стадии дифференцировки. Когда промежуточных стадий отсеквенировано достаточно много, можно отследить путь дифференцировки клетки в транскриптомном пространстве в течение «псевдовремени»[46]. Этот инструментарий помогает изучать механизмы онтогенеза в частности и формирования различий в общем. Сейчас существует множество различных подходов к реконструкции таких траекторий[47].
Применение
Исследования дифференцировки стволовых клеток
Отличия между отдельными клетками — фундаментальная характеристика популяций стволовых клеток, но эти отличия размываются при традиционном анализе ансамблей клеток. Секвенирование РНК одиночных клеток позволяет выявлять эти отличия и обнаруживать различные фенотипы стволовых клеток даже в пределах «однородной» популяции[5].
Так, были выявлены значительные различия между долгоживущими и короткоживущими гематопоэтическими стволовыми клетками мыши и определено, что основной вклад в эти различия вносят гены, отвечающие за клеточный цикл[48][49]. Секвенирование РНК одиночных клеток было применено для изучения лёгких мыши[50] и позволило найти ранее неизвестные маркеры, специфичные для различных подтипов клеток. Были также исследованы нейронные стволовые клетки различных видов и их траектории развития[51]. В другом исследовании было проведено сравнение смен стадий нейронных стволовых клеток у здоровых мышей и мышей, перенёсших ишемию головного мозга[52].
Исследования эмбриогенеза
Процесс эмбрионального развития можно рассматривать как переход от уровня отдельных клеток к уровню организма. Для изучения ранних стадий эмбрионального развития необходимы методы, способные работать с небольшим количеством доступных клеток. С помощью секвенирования РНК одиночных клеток удалось провести общий анализ раннего развития млекопитающих[53][54][55]. Были получены профили экспрессии генов для клеток человека и мыши периода предимплантационного развития[56][57], а также для первичных половых клеток человека в период перехода от стадии миграции к стадии гонад[58]. На клетках мышиных эмбрионов были изучены изменения экспрессии генов в период материнско-зиготического перехода[59][60] (процесс замены зародышем материнских мРНК на свои собственные). Было показано, что в эмбрионе мыши активация зиготического генома происходит на стадии 4 клеток, у человека — между четырёх- и восьмиклеточной стадиями[57]. Для нематоды Caenorhabditis elegans был составлен молекулярный атлас её эмбрионального развития с клеточным разрешением[61].
Анализ тканей
Изучение транскриптома всех клеток ткани даёт возможность узнать больше о иерархии клеточных линий с высокой точностью. Параллельные исследования транскриптомики отдельных клеток селезёнки без предварительного отбора клеток, основанного на заранее выбранных клеточных маркерах, в сочетании с иерархической кластеризацией позволило воссоздать общую структуру взаимоотношений клеточных линий селезёнки[62].
Онкологические исследования
Ткань злокачественной опухоли обычно состоит из нескольких популяций клеток, отличающихся друг от друга функционально и фенотипически. Согласно современным представлениям, процесс развития опухоли может иметь в своей основе не только клональную эволюцию мутировавших клеток исходной ткани, но и иерархическую дифференцировку так называемых раковых стволовых клеток (РСК). Согласно концепции РСК, любое злокачественное новообразование развивается из одной клетки-предшественника популяции РСК, а опухоль устроена иерархически, то есть разные типы раковых клеток обладают разной способностью к делению[63]. Секвенирование РНК одиночных клеток позволяет выявлять отдельные РСК, а также анализировать различные популяции клеток, находящиеся в одной опухоли[63].
Так, недавно были проанализированы транскриптомные профили сотен отдельных опухолевых клеток пяти пациентов с глиобластомой, что позволило выявить дифференциальную экспрессию генов, связанных с онкогенным сигнализированием, пролиферацией, комплементным и иммунным ответом и гипоксией. Также были обнаружены клетки с фенотипами, промежуточными между мезенхимальным и эпителиальным, что не соответствует классической модели эпителиально-мезенхимального перехода с двумя дискретными состояниями клеток. Кроме того, был получен набор генов «стволовости», и клетки также распределялись по непрерывной, а не дискретной шкале уровней экспрессии этих генов, что отражает сложный характер системы стволовых клеток в опухоли[64].
На данный момент существует несколько моделей метастазирования, таких как позднее распространение, ранний сев и самосев, однако до сих пор сложно объяснить ими метастазирование в большинстве видов рака у человека. Трудности заключаются как в упомянутой выше гетерогенности клеток в пределах самой опухоли, так и в сложности анализа ключевых агентов метастазирования — циркулирующих опухолевых клеток(ЦОК): эти клетки исключительно редко встречаются в крови (одна на миллион)[65].
Тем не менее, в недавнем исследовании с помощью секвенирования РНК одиночных клеток удалось выявить три различные генетические подписи в ЦОК, ассоциированные с метастазированием, у пациентов с меланомой[66] . В другом исследовании изучалось распространение отдельных циркулирующих опухолевых клеток и их кластеров в метастатическом раке молочной железы человека, в том числе с использованием мышиных моделей. Было показано, что кластеры имеют повышенный метастатический потенциал по сравнению с отдельными ЦОК, а также что плакоглобин регулирует образование таких кластеров[67]. Исследование отдельных ЦОК метастатического рака поджелудочной железы показало, что эти клетки экспрессируют особые собственные белки внеклеточного матрикса[68]. Подобные результаты позволяют лучше понять функционирование РСК и генетические взаимосвязи между клетками исходной опухоли и метастазов.
Отдельная тема онкологических исследований — приобретение клетками опухоли устойчивости к химиотерапии. Этот процесс также до сих пор плохо изучен для большинства видов рака у человека. В одном из последних исследований были проанализированы транскриптомные профили нескольких сотен отдельных клеток клеточной линии аденокарциномы лёгкого и выявлены новые сигнальные пути, ассоциированные с устойчивостью к определённым компонентам химиотерапии[69]. Исследование ЦОК рака предстательной железы выявило активацию неканонического сигнального пути Wnt, способствующую устойчивости к лекарствам на основе антиандрогена[70].
Исследования альтернативного сплайсинга
Большинство генов эукариот подвержены альтернативному сплайсингу — явлению, позволяющему комбинировать экзоны гена в разных комбинациях, вследствие чего с одного гена появляется возможность производить различные транскрипты и, следовательно, различные белки с потенциально разными функциями. Несмотря на то, что некоторые методы секвенирования РНК одиночных клеток (например, SMART-Seq) имеют близкое к полному покрытие транскриптома, анализ альтернативных изоформ затруднён из-за перечисленных ранее ограничений методов. Например, транскрипты, присутствующие в малом количестве, могут быть не обнаружены из-за неотличимости от биологического шума. Однако, уже разрабатываются модели, учитывающие распределения транскриптов в объединённом множестве отдельно секвенированных клеток[71][72]. Они позволят точнее предсказывать число различных изоформ в отдельных клетках[71].
Иммунология
Секвенирование РНК одиночных клеток может использоваться для эффективного анализа иммунного ответа клеток одной популяции, находящихся в разных условиях. Так, в недавнем исследовании изучалась динамика взаимодействия макрофагов сальмонеллы с клетками-хозяевами c различными модификациями липополисахаридов (основного компонента клеточной стенки)[73]. В другом исследовании изучалась реакция на липополисахариды дендритных клеток костного мозга мышей[74].
См. также
Примечания
- Haque Ashraful, Engel Jessica, Teichmann Sarah A., Lönnberg Tapio. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications (англ.) // Genome Medicine. — 2017. — 18 August (vol. 9, no. 1). — ISSN 1756-994X. — doi:10.1186/s13073-017-0467-4.
- Herr Amy E., Kitamori Takehiko, Landegren Ulf, Kamali-Moghaddam Masood. Next wave advances in single-cell analyses (англ.) // The Analyst. — 2019. — Vol. 144, no. 3. — P. 735—737. — ISSN 0003-2654. — doi:10.1039/c9an90011j.
- Chen Haide, Ye Fang, Guo Guoji. Revolutionizing immunology with single-cell RNA sequencing (англ.) // Cellular & Molecular Immunology. — 2019. — 22 February (vol. 16, no. 3). — P. 242—249. — ISSN 1672-7681. — doi:10.1038/s41423-019-0214-4.
- Kumar Pavithra, Tan Yuqi, Cahan Patrick. Understanding development and stem cells using single cell-based analyses of gene expression (англ.) // Development. — 2017. — 1 January (vol. 144, no. 1). — P. 17—32. — ISSN 0950-1991. — doi:10.1242/dev.133058.
- Svensson Valentine, Vento-Tormo Roser, Teichmann Sarah A. Exponential scaling of single-cell RNA-seq in the past decade (англ.) // Nature Protocols. — 2018. — 1 March (vol. 13, no. 4). — P. 599—604. — ISSN 1754-2189. — doi:10.1038/nprot.2017.149.
- Hedlund Eva, Deng Qiaolin. Single-cell RNA sequencing: Technical advancements and biological applications (англ.) // Molecular Aspects of Medicine. — 2018. — February (vol. 59). — P. 36—46. — ISSN 0098-2997. — doi:10.1016/j.mam.2017.07.003.
- Hwang Byungjin, Lee Ji Hyun, Bang Duhee. Single-cell RNA sequencing technologies and bioinformatics pipelines (англ.) // Experimental & Molecular Medicine. — 2018. — August (vol. 50, no. 8). — ISSN 2092-6413. — doi:10.1038/s12276-018-0071-8.
- Kolodziejczyk Aleksandra A., Kim Jong Kyoung, Svensson Valentine, Marioni John C., Teichmann Sarah A. The Technology and Biology of Single-Cell RNA Sequencing (англ.) // Molecular Cell. — 2015. — May (vol. 58, no. 4). — P. 610—620. — ISSN 1097-2765. — doi:10.1016/j.molcel.2015.04.005.
- Zhang Xiannian, Li Tianqi, Liu Feng, Chen Yaqi, Yao Jiacheng, Li Zeyao, Huang Yanyi, Wang Jianbin. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems (англ.) // Molecular Cell. — 2019. — January (vol. 73, no. 1). — P. 130—142.e5. — ISSN 1097-2765. — doi:10.1016/j.molcel.2018.10.020.
- White A. K., VanInsberghe M., Petriv O. I., Hamidi M., Sikorski D., Marra M. A., Piret J., Aparicio S., Hansen C. L. High-throughput microfluidic single-cell RT-qPCR (англ.) // Proceedings of the National Academy of Sciences. — 2011. — 1 August (vol. 108, no. 34). — P. 13999—14004. — ISSN 0027-8424. — doi:10.1073/pnas.1019446108.
- Sanchez-Freire Veronica, Ebert Antje D, Kalisky Tomer, Quake Stephen R, Wu Joseph C. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns (англ.) // Nature Protocols. — 2012. — 5 April (vol. 7, no. 5). — P. 829—838. — ISSN 1754-2189. — doi:10.1038/nprot.2012.021.
- Everaert Celine, Luypaert Manuel, Maag Jesper L. V., Cheng Quek Xiu, Dinger Marcel E., Hellemans Jan, Mestdagh Pieter. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data (англ.) // Scientific Reports. — 2017. — 8 May (vol. 7, no. 1). — ISSN 2045-2322. — doi:10.1038/s41598-017-01617-3.
- Kameishi Sumako, Umemoto Terumasa, Matsuzaki Yu, Fujita Masako, Okano Teruo, Kato Takashi, Yamato Masayuki. Characterization of rabbit limbal epithelial side population cells using RNA sequencing and single-cell qRT-PCR (англ.) // Biochemical and Biophysical Research Communications. — 2016. — May (vol. 473, no. 3). — P. 704—709. — ISSN 0006-291X. — doi:10.1016/j.bbrc.2015.10.155.
- Ziegenhain Christoph, Vieth Beate, Parekh Swati, Reinius Björn, Guillaumet-Adkins Amy, Smets Martha, Leonhardt Heinrich, Heyn Holger, Hellmann Ines, Enard Wolfgang. Comparative Analysis of Single-Cell RNA Sequencing Methods (англ.) // Molecular Cell. — 2017. — February (vol. 65, no. 4). — P. 631—643.e4. — ISSN 1097-2765. — doi:10.1016/j.molcel.2017.01.023.
- Gao Dan, Jin Feng, Zhou Min, Jiang Yuyang. Recent advances in single cell manipulation and biochemical analysis on microfluidics (англ.) // The Analyst. — 2019. — Vol. 144, no. 3. — P. 766—781. — ISSN 0003-2654. — doi:10.1039/c8an01186a.
- Svec David, Andersson Daniel, Pekny Milos, Sjöback Robert, Kubista Mikael, Ståhlberg Anders. Direct Cell Lysis for Single-Cell Gene Expression Profiling (англ.) // Frontiers in Oncology. — 2013. — Vol. 3. — ISSN 2234-943X. — doi:10.3389/fonc.2013.00274.
- Zhang Yi, Gao Jiaxin, Huang Yanyi, Wang Jianbin. Recent Developments in Single-Cell RNA-Seq of Microorganisms (англ.) // Biophysical Journal. — 2018. — July (vol. 115, no. 2). — P. 173—180. — ISSN 0006-3495. — doi:10.1016/j.bpj.2018.06.008.
- Zajac Pawel, Islam Saiful, Hochgerner Hannah, Lönnerberg Peter, Linnarsson Sten. Base Preferences in Non-Templated Nucleotide Incorporation by MMLV-Derived Reverse Transcriptases (англ.) // PLoS ONE. — 2013. — 31 December (vol. 8, no. 12). — P. e85270. — ISSN 1932-6203. — doi:10.1371/journal.pone.0085270.
- Chen Geng, Ning Baitang, Shi Tieliu. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis (англ.) // Frontiers in Genetics. — 2019. — 5 April (vol. 10). — ISSN 1664-8021. — doi:10.3389/fgene.2019.00317.
- Kim Daehwan, Pertea Geo, Trapnell Cole, Pimentel Harold, Kelley Ryan, Salzberg Steven L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions (англ.) // Genome Biology. — 2013. — Vol. 14, no. 4. — P. R36. — ISSN 1465-6906. — doi:10.1186/gb-2013-14-4-r36.
- Kim Daehwan, Langmead Ben, Salzberg Steven L. HISAT: a fast spliced aligner with low memory requirements (англ.) // Nature Methods. — 2015. — 9 March (vol. 12, no. 4). — P. 357—360. — ISSN 1548-7091. — doi:10.1038/nmeth.3317.
- Büttner Maren, Miao Zhichao, Wolf F. Alexander, Teichmann Sarah A., Theis Fabian J. A test metric for assessing single-cell RNA-seq batch correction (англ.) // Nature Methods. — 2018. — 20 December (vol. 16, no. 1). — P. 43—49. — ISSN 1548-7091. — doi:10.1038/s41592-018-0254-1.
- Bacher Rhonda, Chu Li-Fang, Leng Ning, Gasch Audrey P, Thomson James A, Stewart Ron M, Newton Michael, Kendziorski Christina. SCnorm: robust normalization of single-cell RNA-seq data (англ.) // Nature Methods. — 2017. — 17 April (vol. 14, no. 6). — P. 584—586. — ISSN 1548-7091. — doi:10.1038/nmeth.4263.
- Katayama Shintaro, Töhönen Virpi, Linnarsson Sten, Kere Juha. SAMstrt: statistical test for differential expression in single-cell transcriptome with spike-in normalization (англ.) // Bioinformatics. — 2013. — 31 August (vol. 29, no. 22). — P. 2943—2945. — ISSN 1460-2059. — doi:10.1093/bioinformatics/btt511.
- van Dijk David, Sharma Roshan, Nainys Juozas, Yim Kristina, Kathail Pooja, Carr Ambrose J., Burdziak Cassandra, Moon Kevin R., Chaffer Christine L., Pattabiraman Diwakar, Bierie Brian, Mazutis Linas, Wolf Guy, Krishnaswamy Smita, Pe’er Dana. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion (англ.) // Cell. — 2018. — July (vol. 174, no. 3). — P. 716—729.e27. — ISSN 0092-8674. — doi:10.1016/j.cell.2018.05.061.
- Talwar Divyanshu, Mongia Aanchal, Sengupta Debarka, Majumdar Angshul. AutoImpute: Autoencoder based imputation of single-cell RNA-seq data (англ.) // Scientific Reports. — 2018. — 5 November (vol. 8, no. 1). — ISSN 2045-2322. — doi:10.1038/s41598-018-34688-x.
- Rostom R., Svensson V., Teichmann S. A., Kar G. Computational approaches for interpreting scRNA-seq data. (англ.) // FEBS Letters. — 2017. — August (vol. 591, no. 15). — P. 2213—2225. — doi:10.1002/1873-3468.12684. — PMID 28524227.
- Zhang X., Li T., Liu F., Chen Y., Yao J., Li Z., Huang Y., Wang J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. (англ.) // Molecular Cell. — 2019. — 3 January (vol. 73, no. 1). — P. 130—142. — doi:10.1016/j.molcel.2018.10.020. — PMID 30472192.
- Ntranos Vasilis, Kamath Govinda M., Zhang Jesse M., Pachter Lior, Tse David N. Fast and accurate single-cell RNA-seq analysis by clustering of transcript-compatibility counts (англ.) // Genome Biology. — 2016. — 26 May (vol. 17, no. 1). — ISSN 1474-760X. — doi:10.1186/s13059-016-0970-8.
- Satija Rahul, Farrell Jeffrey A, Gennert David, Schier Alexander F, Regev Aviv. Spatial reconstruction of single-cell gene expression data (англ.) // Nature Biotechnology. — 2015. — 13 April (vol. 33, no. 5). — P. 495—502. — ISSN 1087-0156. — doi:10.1038/nbt.3192.
- Baran Yael, Sebe-Pedros Arnau, Lubling Yaniv, Giladi Amir, Chomsky Elad, Meir Zohar, Hoichman Michael, Lifshitz Aviezer, Tanay Amos. MetaCell: analysis of single cell RNA-seq data using k-NN graph partitions (англ.). — 2018. — 8 October. — doi:10.1101/437665.
- Zhang Jesse M., Fan Jue, Fan H. Christina, Rosenfeld David, Tse David N. An interpretable framework for clustering single-cell RNA-Seq datasets (англ.) // BMC Bioinformatics. — 2018. — 9 March (vol. 19, no. 1). — ISSN 1471-2105. — doi:10.1186/s12859-018-2092-7.
- Tung Po-Yuan, Blischak John D., Hsiao Chiaowen Joyce, Knowles David A., Burnett Jonathan E., Pritchard Jonathan K., Gilad Yoav. Batch effects and the effective design of single-cell gene expression studies (англ.) // Scientific Reports. — 2017. — 3 January (vol. 7, no. 1). — ISSN 2045-2322. — doi:10.1038/srep39921.
- Hicks Stephanie C, Townes F. William, Teng Mingxiang, Irizarry Rafael A. Missing Data and Technical Variability in Single-Cell RNA- Sequencing Experiments (англ.). — 2015. — 25 August. — doi:10.1101/025528.
- Chen Huidong, Albergante Luca, Hsu Jonathan Y., Lareau Caleb A., Lo Bosco Giosuè, Guan Jihong, Zhou Shuigeng, Gorban Alexander N., Bauer Daniel E., Aryee Martin J., Langenau David M., Zinovyev Andrei, Buenrostro Jason D., Yuan Guo-Cheng, Pinello Luca. Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM (англ.) // Nature Communications. — 2019. — 23 April (vol. 10, no. 1). — ISSN 2041-1723. — doi:10.1038/s41467-019-09670-4.
- Lall Snehalika, Sinha Debajyoti, Bandyopadhyay Sanghamitra, Sengupta Debarka. Structure-Aware Principal Component Analysis for Single-Cell RNA-seq Data (англ.) // Journal of Computational Biology. — 2018. — December (vol. 25, no. 12). — P. 1365—1373. — ISSN 1557-8666. — doi:10.1089/cmb.2018.0027.
- Kobak Dmitry, Berens Philipp. The art of using t-SNE for single-cell transcriptomics (англ.). — 2018. — 25 October. — doi:10.1101/453449.
- Wang Bo, Zhu Junjie, Pierson Emma, Ramazzotti Daniele, Batzoglou Serafim. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning (англ.). — 2016. — 9 May. — doi:10.1101/052225.
- Becht Etienne, McInnes Leland, Healy John, Dutertre Charles-Antoine, Kwok Immanuel W H, Ng Lai Guan, Ginhoux Florent, Newell Evan W. Dimensionality reduction for visualizing single-cell data using UMAP (англ.) // Nature Biotechnology. — 2018. — 3 December (vol. 37, no. 1). — P. 38—44. — ISSN 1087-0156. — doi:10.1038/nbt.4314.
- Finak Greg, McDavid Andrew, Yajima Masanao, Deng Jingyuan, Gersuk Vivian, Shalek Alex K., Slichter Chloe K., Miller Hannah W., McElrath M. Juliana, Prlic Martin, Linsley Peter S., Gottardo Raphael. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data (англ.) // Genome Biology. — 2015. — December (vol. 16, no. 1). — ISSN 1474-760X. — doi:10.1186/s13059-015-0844-5.
- Kharchenko Peter V, Silberstein Lev, Scadden David T. Bayesian approach to single-cell differential expression analysis (англ.) // Nature Methods. — 2014. — 18 May (vol. 11, no. 7). — P. 740—742. — ISSN 1548-7091. — doi:10.1038/nmeth.2967.
- Emmert-Streib Frank, Dehmer Matthias, Haibe-Kains Benjamin. Gene regulatory networks and their applications: understanding biological and medical problems in terms of networks (англ.) // Frontiers in Cell and Developmental Biology. — 2014. — 19 August (vol. 2). — ISSN 2296-634X. — doi:10.3389/fcell.2014.00038.
- Kotera Masaaki, Yamanishi Yoshihiro, Moriya Yuki, Kanehisa Minoru, Goto Susumu. GENIES: gene network inference engine based on supervised analysis (англ.) // Nucleic Acids Research. — 2012. — 14 June (vol. 40, no. W1). — P. W162—W167. — ISSN 0305-1048. — doi:10.1093/nar/gks459.
- Shmulevich I., Dougherty E. R., Kim S., Zhang W. Probabilistic Boolean networks: a rule-based uncertainty model for gene regulatory networks (англ.) // Bioinformatics. — 2002. — 1 February (vol. 18, no. 2). — P. 261—274. — ISSN 1367-4803. — doi:10.1093/bioinformatics/18.2.261.
- Zhang Xiujun, Zhao Xing-Ming, He Kun, Lu Le, Cao Yongwei, Liu Jingdong, Hao Jin-Kao, Liu Zhi-Ping, Chen Luonan. Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information (англ.) // Bioinformatics. — 2011. — 15 November (vol. 28, no. 1). — P. 98—104. — ISSN 1460-2059. — doi:10.1093/bioinformatics/btr626.
- Griffiths Jonathan A, Scialdone Antonio, Marioni John C. Using single‐cell genomics to understand developmental processes and cell fate decisions (англ.) // Molecular Systems Biology. — 2018. — April (vol. 14, no. 4). — ISSN 1744-4292. — doi:10.15252/msb.20178046.
- Saelens Wouter, Cannoodt Robrecht, Todorov Helena, Saeys Yvan. A comparison of single-cell trajectory inference methods (англ.) // Nature Biotechnology. — 2019. — 1 April (vol. 37, no. 5). — P. 547—554. — ISSN 1087-0156. — doi:10.1038/s41587-019-0071-9.
- Kowalczyk Monika S., Tirosh Itay, Heckl Dirk, Rao Tata Nageswara, Dixit Atray, Haas Brian J., Schneider Rebekka K., Wagers Amy J., Ebert Benjamin L., Regev Aviv. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells (англ.) // Genome Research. — 2015. — 1 October (vol. 25, no. 12). — P. 1860—1872. — ISSN 1088-9051. — doi:10.1101/gr.192237.115.
- Tsang Jason C. H., Yu Yong, Burke Shannon, Buettner Florian, Wang Cui, Kolodziejczyk Aleksandra A., Teichmann Sarah A., Lu Liming, Liu Pentao. Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells (англ.) // Genome Biology. — 2015. — 21 September (vol. 16, no. 1). — ISSN 1474-760X. — doi:10.1186/s13059-015-0739-5.
- Treutlein Barbara, Brownfield Doug G., Wu Angela R., Neff Norma F., Mantalas Gary L., Espinoza F. Hernan, Desai Tushar J., Krasnow Mark A., Quake Stephen R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq (англ.) // Nature. — 2014. — 13 April (vol. 509, no. 7500). — P. 371—375. — ISSN 0028-0836. — doi:10.1038/nature13173.
- Shin Jaehoon, Berg Daniel A., Zhu Yunhua, Shin Joseph Y., Song Juan, Bonaguidi Michael A., Enikolopov Grigori, Nauen David W., Christian Kimberly M., Ming Guo-li, Song Hongjun. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis (англ.) // Cell Stem Cell. — 2015. — September (vol. 17, no. 3). — P. 360—372. — ISSN 1934-5909. — doi:10.1016/j.stem.2015.07.013.
- Llorens-Bobadilla Enric, Zhao Sheng, Baser Avni, Saiz-Castro Gonzalo, Zwadlo Klara, Martin-Villalba Ana. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury (англ.) // Cell Stem Cell. — 2015. — September (vol. 17, no. 3). — P. 329—340. — ISSN 1934-5909. — doi:10.1016/j.stem.2015.07.002.
- Deng Q., Ramskold D., Reinius B., Sandberg R. Single-Cell RNA-Seq Reveals Dynamic, Random Monoallelic Gene Expression in Mammalian Cells (англ.) // Science. — 2014. — 9 January (vol. 343, no. 6167). — P. 193—196. — ISSN 0036-8075. — doi:10.1126/science.1245316.
- Xue Zhigang, Huang Kevin, Cai Chaochao, Cai Lingbo, Jiang Chun-yan, Feng Yun, Liu Zhenshan, Zeng Qiao, Cheng Liming, Sun Yi E., Liu Jia-yin, Horvath Steve, Fan Guoping. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing (англ.) // Nature. — 2013. — 28 July (vol. 500, no. 7464). — P. 593—597. — ISSN 0028-0836. — doi:10.1038/nature12364.
- Tang Fuchou, Barbacioru Catalin, Bao Siqin, Lee Caroline, Nordman Ellen, Wang Xiaohui, Lao Kaiqin, Surani M. Azim. Tracing the Derivation of Embryonic Stem Cells from the Inner Cell Mass by Single-Cell RNA-Seq Analysis (англ.) // Cell Stem Cell. — 2010. — May (vol. 6, no. 5). — P. 468—478. — ISSN 1934-5909. — doi:10.1016/j.stem.2010.03.015.
- Tang Fuchou, Barbacioru Catalin, Nordman Ellen, Bao Siqin, Lee Caroline, Wang Xiaohui, Tuch Brian B., Heard Edith, Lao Kaiqin, Surani M. Azim. Deterministic and Stochastic Allele Specific Gene Expression in Single Mouse Blastomeres (англ.) // PLoS ONE. — 2011. — 23 June (vol. 6, no. 6). — P. e21208. — ISSN 1932-6203. — doi:10.1371/journal.pone.0021208.
- Yan Liying, Yang Mingyu, Guo Hongshan, Yang Lu, Wu Jun, Li Rong, Liu Ping, Lian Ying, Zheng Xiaoying, Yan Jie, Huang Jin, Li Ming, Wu Xinglong, Wen Lu, Lao Kaiqin, Li Ruiqiang, Qiao Jie, Tang Fuchou. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells (англ.) // Nature Structural & Molecular Biology. — 2013. — 11 August (vol. 20, no. 9). — P. 1131—1139. — ISSN 1545-9993. — doi:10.1038/nsmb.2660.
- Guo Fan, Yan Liying, Guo Hongshan, Li Lin, Hu Boqiang, Zhao Yangyu, Yong Jun, Hu Yuqiong, Wang Xiaoye, Wei Yuan, Wang Wei, Li Rong, Yan Jie, Zhi Xu, Zhang Yan, Jin Hongyan, Zhang Wenxin, Hou Yu, Zhu Ping, Li Jingyun, Zhang Ling, Liu Sirui, Ren Yixin, Zhu Xiaohui, Wen Lu, Gao Yi Qin, Tang Fuchou, Qiao Jie. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells (англ.) // Cell. — 2015. — June (vol. 161, no. 6). — P. 1437—1452. — ISSN 0092-8674. — doi:10.1016/j.cell.2015.05.015.
- Biase Fernando H., Cao Xiaoyi, Zhong Sheng. Cell fate inclination within 2-cell and 4-cell mouse embryos revealed by single-cell RNA sequencing (англ.) // Genome Research. — 2014. — 5 August (vol. 24, no. 11). — P. 1787—1796. — ISSN 1088-9051. — doi:10.1101/gr.177725.114.
- Shi Junchao, Chen Qi, Li Xin, Zheng Xiudeng, Zhang Ying, Qiao Jie, Tang Fuchou, Tao Yi, Zhou Qi, Duan Enkui. Dynamic transcriptional symmetry-breaking in pre-implantation mammalian embryo development revealed by single-cell RNA-seq (англ.) // Development. — 2015. — 22 September (vol. 142, no. 20). — P. 3468—3477. — ISSN 0950-1991. — doi:10.1242/dev.123950.
- Packer Jonathan S., Zhu Qin, Huynh Chau, Sivaramakrishnan Priya, Preston Elicia, Dueck Hannah, Stefanik Derek, Tan Kai, Trapnell Cole, Kim Junhyong, Waterston Robert H., Murray John I. A lineage-resolved molecular atlas of C. elegans embryogenesis at single cell resolution (англ.). — 2019. — 1 March. — doi:10.1101/565549.
- Jaitin D. A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A., Amit I. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. (англ.) // Science (New York, N.Y.). — 2014. — 14 February (vol. 343, no. 6172). — P. 776—779. — doi:10.1126/science.1247651. — PMID 24531970.
- В центре внимания раковые стволовые клетки. elementy.ru.
- Patel A. P., Tirosh I., Trombetta J. J., Shalek A. K., Gillespie S. M., Wakimoto H., Cahill D. P., Nahed B. V., Curry W. T., Martuza R. L., Louis D. N., Rozenblatt-Rosen O., Suva M. L., Regev A., Bernstein B. E. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma (англ.) // Science. — 2014. — 12 June (vol. 344, no. 6190). — P. 1396—1401. — ISSN 0036-8075. — doi:10.1126/science.1254257.
- Miller MC, Doyle GV, Terstappen LW (2010). “Significance of Circulating Tumor Cells Detected by the CellSearch System in Patients with Metastatic Breast Colorectal and Prostate Cancer”. J Oncol [англ.]. 2010: 1—8. DOI:10.1155/2010/617421. PMC 2793426. PMID 20016752.
- Ramsköld D., Luo S., Wang Y. C., Li R., Deng Q., Faridani O. R., Daniels G. A., Khrebtukova I., Loring J. F., Laurent L. C., Schroth G. P., Sandberg R. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. (англ.) // Nature Biotechnology. — 2012. — August (vol. 30, no. 8). — P. 777—782. — doi:10.1038/nbt.2282. — PMID 22820318.
- Aceto Nicola, Bardia Aditya, Miyamoto David T., Donaldson Maria C., Wittner Ben S., Spencer Joel A., Yu Min, Pely Adam, Engstrom Amanda, Zhu Huili, Brannigan Brian W., Kapur Ravi, Stott Shannon L., Shioda Toshi, Ramaswamy Sridhar, Ting David T., Lin Charles P., Toner Mehmet, Haber Daniel A., Maheswaran Shyamala. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis (англ.) // Cell. — 2014. — August (vol. 158, no. 5). — P. 1110—1122. — ISSN 0092-8674. — doi:10.1016/j.cell.2014.07.013.
- Ting D. T., Wittner B. S., Ligorio M., Vincent Jordan N., Shah A. M., Miyamoto D. T., Aceto N., Bersani F., Brannigan B. W., Xega K., Ciciliano J. C., Zhu H., MacKenzie O. C., Trautwein J., Arora K. S., Shahid M., Ellis H. L., Qu N., Bardeesy N., Rivera M. N., Deshpande V., Ferrone C. R., Kapur R., Ramaswamy S., Shioda T., Toner M., Maheswaran S., Haber D. A. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. (англ.) // Cell Reports. — 2014. — 25 September (vol. 8, no. 6). — P. 1905—1918. — doi:10.1016/j.celrep.2014.08.029. — PMID 25242334.
- Suzuki Ayako, Matsushima Koutatsu, Makinoshima Hideki, Sugano Sumio, Kohno Takashi, Tsuchihara Katsuya, Suzuki Yutaka. Single-cell analysis of lung adenocarcinoma cell lines reveals diverse expression patterns of individual cells invoked by a molecular target drug treatment (англ.) // Genome Biology. — 2015. — 3 April (vol. 16, no. 1). — ISSN 1474-760X. — doi:10.1186/s13059-015-0636-y.
- Miyamoto D. T., Zheng Y., Wittner B. S., Lee R. J., Zhu H., Broderick K. T., Desai R., Fox D. B., Brannigan B. W., Trautwein J., Arora K. S., Desai N., Dahl D. M., Sequist L. V., Smith M. R., Kapur R., Wu C.-L., Shioda T., Ramaswamy S., Ting D. T., Toner M., Maheswaran S., Haber D. A. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance (англ.) // Science. — 2015. — 17 September (vol. 349, no. 6254). — P. 1351—1356. — ISSN 0036-8075. — doi:10.1126/science.aab0917.
- Welch Joshua D., Hu Yin, Prins Jan F. Robust detection of alternative splicing in a population of single cells (англ.) // Nucleic Acids Research. — 2016. — 5 January (vol. 44, no. 8). — P. e73—e73. — ISSN 0305-1048. — doi:10.1093/nar/gkv1525.
- Marinov G. K., Williams B. A., McCue K., Schroth G. P., Gertz J., Myers R. M., Wold B. J. From single-cell to cell-pool transcriptomes: Stochasticity in gene expression and RNA splicing (англ.) // Genome Research. — 2013. — 3 December (vol. 24, no. 3). — P. 496—510. — ISSN 1088-9051. — doi:10.1101/gr.161034.113.
- Avraham R., Haseley N., Brown D., Penaranda C., Jijon H. B., Trombetta J. J., Satija R., Shalek A. K., Xavier R. J., Regev A., Hung D. T. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. (англ.) // Cell. — 2015. — 10 September (vol. 162, no. 6). — P. 1309—1321. — doi:10.1016/j.cell.2015.08.027. — PMID 26343579.
- Shalek Alex K., Satija Rahul, Adiconis Xian, Gertner Rona S., Gaublomme Jellert T., Raychowdhury Raktima, Schwartz Schraga, Yosef Nir, Malboeuf Christine, Lu Diana, Trombetta John J., Gennert Dave, Gnirke Andreas, Goren Alon, Hacohen Nir, Levin Joshua Z., Park Hongkun, Regev Aviv. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells (англ.) // Nature. — 2013. — 19 May (vol. 498, no. 7453). — P. 236—240. — ISSN 0028-0836. — doi:10.1038/nature12172.
Ссылки
.svg.png.webp) Analysis of single cell RNA-seq data (англ.): плей-лист (17 видео) от Bioinformatics Training — University of Cambridge
Analysis of single cell RNA-seq data (англ.): плей-лист (17 видео) от Bioinformatics Training — University of Cambridge- Транскриптомика: практические методы и применяемые алгоритмы А. Предеус, Институт биоинформатики
- Транскриптомика: анализ данных RNA-seq П. Мазин, Факультет биоинженерии и биоинформатики МГУ
- RNA-seqlopedia от University of Oregon (англ.). rnaseq.uoregon.edu. Дата обращения: 29 апреля 2017.
- Анализ данных single cell Константин Зайцев, Washington University in St.Louis, Институт биоинформатики
- Многосторонность и ограничения анализа одиночных клеток Константин Оконечников, Институт биоинформатики
- Изучение развития мозга на уровне одиночных клеток Константин Оконечников, Институт биоинформатики