Секвенирование ДНК одиночных клеток
Секвени́рование ДНК одино́чных кле́ток (англ. Single-cell DNA sequencing) — подход, позволяющий получить данные о последовательности ДНК отдельной клетки с помощью секвенирования и, следовательно, выявлять различия между отдельными клетками одноклеточных организмов, органов, тканей и клеточных субпопуляций многоклеточных организмов. Подход позволяет анализировать функциональные особенности клетки в контексте микроокружения. Секвенирование генома единичной клетки включает несколько шагов: выделение одной клетки, полногеномная амплификация, создание библиотек и секвенирование ДНК с использованием методов секвенирования нового поколения.
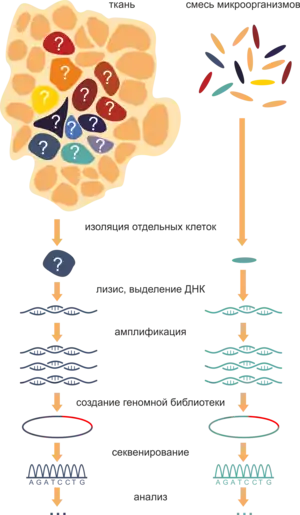
С появлением разнообразных методов секвенирования возникла возможность устанавливать последовательность геномной ДНК. Однако большинство данных на текущий момент получены при секвенировании образцов геномной ДНК, выделенных из популяций микроорганизмов или клеточных субпопуляций многоклеточных организмов[1]. Однако известно, что разнообразие внутри обеих групп может быть существенным, так как сами клетки вносят разный вклад в существование популяции или организма.
Секвенирование генома единичной клетки позволяет перевести изучение генома на клеточный уровень. Сегодня это помогает решать такие задачи, как de novo секвенирование некультивируемых микроорганизмов[2], изучение генетического мозаицизма в нормальных и патологических случаях[3], выявление и изучение вклада клеточных субпопуляций опухолей в развитие рака и возникновение устойчивости к лечению[4].
Технологические задачи
Перед секвенированием ДНК одиночных клеток стоят задачи физического выделения отдельных клеток, выбора метода амплификации с наименьшей вероятностью внесения ошибок для получения достаточного количества материала и выбора способа секвенирования[5][6].
Изолирование отдельных клеток
Первым шагом в изолировании клеток является создание суспензии жизнеспособных клеток, не связанных друг с другом. Целью изолирования может быть как случайный выбор клеток для создания репрезентативной выборочной совокупности при анализе состава субпопуляций, так и целенаправленный поиск определенных клеток. При исследовании твердых тканей необходима предварительная механическая или химическая диссоциация образца, при этом условия диссоциации должны одинаково действовать на все субпопуляции клеток тканей. Это необходимо для создания несмещенной относительно исходного набора клеток выборки, где сохраняется изначальная представленность клеток, что может быть важно для анализа состава субпопуляций. Стоит учитывать, что условия диссоциации нормальных и нездоровых тканей могут различаться, поэтому на данном этапе важно подобрать соответствующие условия. Работа с цельными образцами тканей также возможна, например, с помощью лазерной захватывающей микродиссекции[7].
После получения суспензии можно приступать к изолированию клеток методами серийного разведения[8], микропипетирования[9], разведения в микроячейках[10], с использованием оптического пинцета. Метод проточной флуоресцентной цитометрии может использоваться для отделения клеток с определенными флуоресцентными свойствами, которые могут быть как естественными, так и введены экспериментатором. Большое развитие в последнее время получили автоматизированные методы микроманипуляции[11][12], в том числе изолирование клеток на чипах с использованием технологий микрофлюидики[13]; взятие нанобиопсий уже позволяет исследовать ДНК отдельных органелл[14]. Изолированные клетки впоследствии подвергаются лизису.
Полногеномная амплификация
Следующий шаг — полногеномная амплификация (англ. whole genome amplification, WGA), — служит для наработки такого количества ДНК, которого достаточно для детекции сигнала и его выделения из шума в дальнейшем при секвенировании. При этом желательно минимизировать внесение таких артефактов, как предпочтительная амплификация простых последовательностей, введение случайных мутаций и формирование химерных последовательностей. За последнее время появился набор возможностей для решения этой задачи. Использование ПЦР не оправдало себя ввиду, например, повышенной частоты введения ошибок термостабильными полимеразами. Поэтому наибольшего распространения добились изотермические и гибридные методы, такие как метод амплификации со множественным замещением цепи (англ. Multiple displacement amplification, MDA) и амплификации со множественным выпрямлением и выпетливанием (англ. Multiple Annealing and Looping Based Amplification Cycles, MALBAC)[15].
MDA
MDA позволяет быстро амплифицировать ДНК, не задействуя ПЦР. Метод основан на использовании фаговой полимеразы phi29, которая характеризуется повышенной процессивностью (может синтезировать участки длиной свыше 10 килобаз без диссоциации) и низкой частотой ошибок (1 на 106−107 пар оснований). Реакция происходит следующим образом: гексамерные праймеры отжигаются на матрице, элонгируются посредством полимеразы; когда фермент встречает другой праймер (который также элонгируется), то он вытесняет (замещает) его и продолжает свой путь по матрице. Замещенный новосинтезированный участок служит местом посадки новых праймеров и становится матрицей. Таким образом формируется ветвистое дерево, где синтез происходит на каждой ветви. В конце процедуры полимераза ингибируется, добавляется нуклеаза S1 для отщепления ветвей в местах ветвления и ДНК-полимераза I для достраивания образующихся одноцепочечных участков[15].
Метод имеет ряд проблем, таких как потеря аллелей, предпочтительная амплификация и взаимодействия между праймерами. Первая проблема возникает вследствие случайной амплификации только одного из аллелей в гетерозиготах, в результате чего гетерозиготы неверно определяются как гомозиготы. Из-за высокой частоты проявления этого эффекта (0 — 60 %) уменьшается точность генотипирования. Вторая проблема заключается в сверхамплификации одного аллеля по сравнению с другими. Взаимодействия между гексамерными праймерами происходят вследствие случайного характера последовательностей; их можно значительно уменьшить, введя ограничения при синтезе этих праймеров[15].
MALBAC
MALBAC — гибридный линейный метод полногеномной амплификации. Основа метода — специальные праймеры: они имеют длину 35 нуклеотидов, 27 из которых одинаковы во всех праймерах (GTG AGT GAT GGT TGA GGT AGT GTG GAG), а 8 оставшихся нуклеотидов варьируются. Весь процесс амплификации описывается следующим образом[9]:
- Плавление (94 °С) двухцепочечной ДНК с образованием одноцепочечных фрагментов.
- Охлаждение (0 °С), добавление праймеров и полимеразы.
- Отжиг праймеров в случайных местах на матрице. ДНК-полимераза Bst удлиняет праймеры с образованием полуампликона при 64 °С. Все встречные праймеры смещаются с матрицы.
- Плавление (94 °С), отделение полуампликона от матрицы.
- Охлаждение (0 °С), добавление праймеров и полимеразы. Праймеры эффективно связываются и с матрицей, и с полуампликоном.
- ДНК-полимераза Bst удлиняет праймеры при 64 °С. На исходной матрице синтезируются полуампликоны, на полуампликонах, полученных ранее, синтезируются полные ампликоны.
- Плавление (94 °С).
- Образование петель (58 °С): у полных ампликонов 3' и 5' концы комплементарны друг другу и образуют петлю, не допуская использования полного ампликона в качестве матрицы.
- Повторение шагов 5—8 пять раз.
- ПЦР с использованием 27 общих нуклеотидов в качестве праймеров для амплификации только полных ампликонов[9].
Преимуществом метода является уменьшение шума, связанного с экспоненциальным характером ПЦР амплификации, благодаря введению предварительной квази-линейной амплификации. Это позволило увеличить покрытие генома (доля генома, покрытая хотя бы одним ридом), уменьшить вероятность потери аллелей и однонуклеотидных полиморфизмов (SNPs). Помимо этого, на вход требуется очень небольшое количество исходной ДНК, однако любое загрязнение образцов способно значительно повлиять на результаты секвенирования[9].
Недостатком является то, что для избавления от ложно-положительных результатов необходимо сравнивать результаты секвенирования 2—3 клеток как из той же, так и из иной клеточных линий[9]. При этом может теряться часть полиморфизмов, так как клетки, принадлежащие одной клеточной линии, все же имеют некоторые различия в геноме. Кроме того, используемая ДНК-полимераза bst имеет высокую частоту ошибок (1 на 105 оснований)[16].
Сравнение методов полногеномной амплификации
В последнее время было проведено несколько исследований, посвященных сравнению этих методов[17][18][19]. Результатом одного из исследований стал вывод о том, что MDA позволяет получить большее покрытие, чем MALBAC (84 % и 52 % соответственно), что позволяет более точно определять однонуклеотидные полиморфизмы[17]. Однако MALBAC обеспечивает более равномерное покрытие и поэтому дает возможность более точно выявлять вариации числа копий (CNVs)[17]. Интересно, что при секвенировании некоторых клеток уровень детекции вариаций числа копий методом MDA был сопоставим с MALBAC[17]. Другие авторы также подтверждают разницу в покрытии между MDA и MALBAC (84 % и 72 %) и сравнительно более высокую равномерность покрытия MALBAC (коэффициент вариации 0,10 против 0,21 у MDA)[18]. Показано, что MDA дает меньше ложно-положительных результатов, но число ложно-отрицательных результатов меняется от эксперимента к эксперименту[18]. MALBAC дает меньшую частоту потери аллелей (21 %), однако и покрытие его меньше, чем у MDA[18]. В целом не ясно, какой приводит к меньшему количеству ложно-отрицательных результатов, так как MDA покрывает большую часть генома, но при этом теряет больше аллелей из-за предпочтительной амплификации только одного из аллелей в гетерозиготе[15][18].
Таким образом, MDA и MALBAC имеют набор преимуществ и недостатков, и выбор должен зависеть от поставленной задачи.
Создание библиотеки
После амплификации можно приступать к приготовлению библиотек с помощью коммерческих наборов. Здесь возможно несколько вариантов: выбор определенного локуса, выбор экзома или всего генома для дальнейшего секвенирования. Каждый из этих вариантов предполагает определенные значения покрытия, склонности к ошибкам и стоимости[20]. Выбор небольших участков позволяет сфокусироваться на областях, вносящих наибольший биологический вклад в работу изучаемой системы. При этом уменьшается цена исследования и вероятность внесения ошибок при подготовке проб. Использование референсного генома позволяет уменьшить ложно-положительные результаты, хотя и ограничивает определяемые однонуклеотидные полиморфизмы теми, что присутствуют в референсном геноме. Секвенирование экзома позволяет выделить уникальные особенности клеток, однако с ростом длины секвенируемого участка растет вероятность внесения ошибок в ходе амплификации. Использование всего генома позволяет выявить некодирующие и структурные участки, однако стоимость исследования резко возрастает, что затрудняет полногеномное секвенирование многих клеток[20].
ДНК из созданных тем или иным способом библиотек используется в секвенировании одним из существующих методов.
Обработка данных
Распространенные ошибки
Большинство артефактов секвенирования возникают при подготовке образцов: изоляция клеток, загрязнение геномной ДНК, амплификация и создание библиотек, так как все эти шаги привносят дополнительные ошибки, приводят к потере покрытия, уменьшению однородности покрытия, смещениям в выборке при предпочтительном отборе определенных групп клеток и амплификации определенных последовательностей ДНК, являются причиной потери аллелей в гетерозиготных позициях. Следует также принимать во внимание клеточные линии, на которых проводится оптимизация всех стадий секвенирования: не все клетки диплоидны, есть и гаплоидные, и анеуплоидные популяции, и их плоидность может значительно влиять на эксперимент[4]. Препятствием на пути сравнения различных результатов в этой области подчас является отсутствие информации об общем количестве оцененных клеток и мере оценки качества секвенирования в конкретных работах[20].
Однонуклеотидные полиморфизмы
Однонуклеотидные полиморфизмы, согласно проекту 1000 геномов, вносят наибольшее разнообразие в геном человека[21]: на карте гаплотипа подтверждено 38 млн однонуклеотидных полиморфизмов, 1,4 млн вставок/делеций и более 14 тыс крупных делеций[21]. Также предполагается, что многие комплексные заболевания, такие как болезнь Альцгеймера[22], различные виды рака[23], аутоиммунные заболевания[24] могут быть связаны именно с наличием полиморфизмов.
Сегодня поиск полиморфизмов в данных секвенирования отдельных клеток опирается на те же алгоритмы, что и анализ результатов обычного секвенирования: GATK[25], SNPdetector[26], SOAPsnp[27], VarScan[28]. Однако есть отличия между секвенированием популяции клеток и секвенированием отдельных клеток: в последнем случае меньше покрытие генома и выше уровень ложно-положительных результатов.
Вариация числа копий участков ДНК
Вариации числа копий фрагментов ДНК приводят к ненормальному числу копий этих фрагментов; разнообразие этого типа генетического полиморфизма также влияет на здоровье людей[29][30]. Некоторые исследования подчеркивают их связь с развитием опухолей[31], аутоиммунных заболеваний[24], аутизма[32] и др. Здесь, как и при поиске однонуклеотидных полиморфизмов, используются в основном те же алгоритмы, что и для обычного секвенирования: CNV-seq[33], PenCNV[34], CNAseg[35], ReadDepth[36], и cn.MOPS[37]. Для того, чтобы учитывать вносимый шум, необходимо произвести анализ влияния методов амплификации на появление и исчезновение вариаций числа копий ДНК[38].
Сравнительный анализ клеток
Одной из стратегий кластеризации клеток исходя из геномных данных является введение функции расстояний, которая обеспечивает количественную оценку различий между парами образцов[39]. В данном случае Мера Жаккара считается наиболее подходящей ввиду бинарной природы генетических данных (см. ниже)[40]. Альтернативой основанным на функции расстояния методам является основанная на модели кластеризация, предполагающая вероятностный подход: вместо «жёстких» расстояний вводятся «мягкие» вероятности происхождения клеток от различных клонов.
Представив данные секвенирования единичных клеток в качестве матрицы, где по вертикали отмечены интересующие нас мутации, а по горизонтали — клетки, заполняем ее 0 и 1 в зависимости от присутствия конкретной мутации в конкретной клетке. Если исследуется опухоль, то для нее с течением времени характерны экспансия одних клонов и исчезновение других[41]. При этом мы не знаем, сколько каких клонов присутствует, и предполагаем, что часть данных потеряна в ходе подготовки проб.
Параметры модели, такие как вероятность клетки произойти от определенного клона, а также уровень ложно-отрицательных результатов, могут быть оценены посредством алгоритма максимизации ожидания[42]. Тогда проблема определения числа клонов сводится к выбору статистической модели, которая наилучшим образом описывает данные секвенирования; оценка проводится с помощью информационных критериев Байеса и Акаике[43]. Существует и гибридный подход, позволяющий осуществлять первоначальную кластеризацию с помощью функции расстояния, что увеличивает скорость основанной на модели кластеризации, требующей больших вычислительных мощностей[44]. Основываясь на результатах кластеризации, строится профиль консенсусных клональных мутаций[45]. По нему с помощью различных методов построения деревьев можно выявлять взаимоотношения между разными клонами. Так, например, можно продемонстрировать эволюционную историю опухоли[45].
Достижения
Клональная эволюция клеток рака груди
Анализ паттернов мутаций (вставки, делеции, однонуклеотидные замены, вариации числа копий генов) различных популяций клеток рака груди позволил выявить как набор мутаций, характерных для каждой из популяций (клональные мутации), так и тех, что встречались в нескольких клетках (субклональные мутации). Данные были получены с помощью секвенирования экзомов отдельных клеток, проверены методом глубокого секвенирования. В исследовании использовались клетки анеуплоидных популяций ERBC (ER+/PR+/Her2-) и TNBC (ER-/PR-/Her2-), отличающихся по наличию определенных рецепторов (ER/PR/Her2) на поверхности мембраны, а также нормальные диплоидные клетки. Результатом стало выявление значительно большего количества клональных мутаций в популяции TNBC по сравнению с ERBC и нормальными клетками. В популяции клеток TNBC показано существование трех субпопуляций раковых клеток, найденных по паттернам субклональных мутаций. Получены доказательства того, что TNBC обладает большей частотой возникновения мутаций, и их накопление может происходить не только из-за ошибок при ускоренной пролиферации[4].
Пока неясно, как именно возникает устойчивость опухолей к химиотерапии. Либо в популяции уже есть редко встречающиеся устойчивые клетки, либо ответ возникает спонтанно после действия лекарств. Кроме того, не всегда ясно, за счет чего накапливаются мутации: либо это ускоренная скорость мутаций, как в случае TNBC, либо это накопление мутаций с обычной скоростью, но в большом количестве вследствие ускорения пролиферации[4].
Перспективы
На данный момент основную проблему представляет наличие шага амплификации геномной ДНК, ответственного за внесение наибольшего числа артефактов. Требования к количеству ДНК при приготовлении библиотек все уменьшаются, и уже было продемонстрировано прямое создание библиотек из выделенной ДНК[46][47]. Более того, была показана возможность вовсе обходиться без библиотек, подавая на секвенирование выделенную из клетки ДНК[48]. Существует также возможность выявления эпигенетической информации, такой как поиск паттернов метилирования[49][50] и захват конформационного состояния хромосом[51]. Сегодня ученые обычно оперируют десятками-сотнями клеток, но развитие автоматических платформ для захвата клеток, амплификации ДНК и приготовления библиотек существенно увеличит масштабы и доступность анализа отдельных клеток, позволяя проводить более крупные эксперименты в короткий срок[52].
Применение метода секвенирования ДНК отдельных клеток вместе с эпигеномными и транскриптомными исследованиями позволит точно классифицировать клетки и дополнить существующий взгляд на клеточные популяции. Также станет возможным установление взаимоотношений между последовательностью генома, эпигенетическим статусом и экспрессией генов, определение функциональных возможностей клеток[52].
См. также
Примечания
- Tringe S. G., von Mering C., Kobayashi A., Salamov A. A., Chen K., Chang H. W., Podar M., Short J. M., Mathur E. J., Detter J. C., Bork P., Hugenholtz P., Rubin E. M. Comparative metagenomics of microbial communities. (англ.) // Science (New York, N.Y.). — 2005. — Vol. 308, no. 5721. — P. 554—557. — doi:10.1126/science.1107851. — PMID 15845853.
- Marcy Y., Ouverney C., Bik E. M., Lösekann T., Ivanova N., Martin H. G., Szeto E., Platt D., Hugenholtz P., Relman D. A., Quake S. R. Dissecting biological "dark matter" with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2007. — Vol. 104, no. 29. — P. 11889—11894. — doi:10.1073/pnas.0704662104. — PMID 17620602.
- McConnell M. J., Lindberg M. R., Brennand K. J., Piper J. C., Voet T., Cowing-Zitron C., Shumilina S., Lasken R. S., Vermeesch J. R., Hall I. M., Gage F. H. Mosaic copy number variation in human neurons. (англ.) // Science (New York, N.Y.). — 2013. — Vol. 342, no. 6158. — P. 632—637. — doi:10.1126/science.1243472. — PMID 24179226.
- Wang Y., Waters J., Leung M. L., Unruh A., Roh W., Shi X., Chen K., Scheet P., Vattathil S., Liang H., Multani A., Zhang H., Zhao R., Michor F., Meric-Bernstam F., Navin N. E. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. (англ.) // Nature. — 2014. — Vol. 512, no. 7513. — P. 155—160. — doi:10.1038/nature13600. — PMID 25079324.
- Wen L., Tang F. Single-cell sequencing in stem cell biology. (англ.) // Genome biology. — 2016. — Vol. 17, no. 1. — P. 71. — doi:10.1186/s13059-016-0941-0. — PMID 27083874.
- Bacher R., Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. (англ.) // Genome biology. — 2016. — Vol. 17, no. 1. — P. 63. — doi:10.1186/s13059-016-0927-y. — PMID 27052890.
- Emmert-Buck M. R., Bonner R. F., Smith P. D., Chuaqui R. F., Zhuang Z., Goldstein S. R., Weiss R. A., Liotta L. A. Laser capture microdissection. (англ.) // Science (New York, N.Y.). — 1996. — Vol. 274, no. 5289. — P. 998—1001. — PMID 8875945.
- HAM R. G. CLONAL GROWTH OF MAMMALIAN CELLS IN A CHEMICALLY DEFINED, SYNTHETIC MEDIUM. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 1965. — Vol. 53. — P. 288—293. — PMID 14294058.
- Zong C., Lu S., Chapman A. R., Xie X. S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. (англ.) // Science (New York, N.Y.). — 2012. — Vol. 338, no. 6114. — P. 1622—1626. — doi:10.1126/science.1229164. — PMID 23258894.
- Gole J., Gore A., Richards A., Chiu Y. J., Fung H. L., Bushman D., Chiang H. I., Chun J., Lo Y. H., Zhang K. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. (англ.) // Nature biotechnology. — 2013. — Vol. 31, no. 12. — P. 1126—1132. — doi:10.1038/nbt.2720. — PMID 24213699.
- White A. K., VanInsberghe M., Petriv O. I., Hamidi M., Sikorski D., Marra M. A., Piret J., Aparicio S., Hansen C. L. High-throughput microfluidic single-cell RT-qPCR. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2011. — Vol. 108, no. 34. — P. 13999—14004. — doi:10.1073/pnas.1019446108. — PMID 21808033.
- Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M., Trombetta J. J., Weitz D. A., Sanes J. R., Shalek A. K., Regev A., McCarroll S. A. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. (англ.) // Cell. — 2015. — Vol. 161, no. 5. — P. 1202—1214. — doi:10.1016/j.cell.2015.05.002. — PMID 26000488.
- Thompson A. M., Paguirigan A. L., Kreutz J. E., Radich J. P., Chiu D. T. Microfluidics for single-cell genetic analysis. (англ.) // Lab on a chip. — 2014. — Vol. 14, no. 17. — P. 3135—3142. — doi:10.1039/c4lc00175c. — PMID 24789374.
- Actis P., Maalouf M. M., Kim H. J., Lohith A., Vilozny B., Seger R. A., Pourmand N. Compartmental genomics in living cells revealed by single-cell nanobiopsy. (англ.) // ACS nano. — 2014. — Vol. 8, no. 1. — P. 546—553. — doi:10.1021/nn405097u. — PMID 24279711.
- Paez J. G., Lin M., Beroukhim R., Lee J. C., Zhao X., Richter D. J., Gabriel S., Herman P., Sasaki H., Altshuler D., Li C., Meyerson M., Sellers W. R. Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. (англ.) // Nucleic acids research. — 2004. — Vol. 32, no. 9. — P. e71. — doi:10.1093/nar/gnh069. — PMID 15150323.
- Ausubel, F.M. et al. Vol. I., John Wiley & Songs, Inc. CURRENT PROTOCOLS IN MOLECULAR BIOLOGY. — 1995.
- Hou Y., Wu K., Shi X., Li F., Song L., Wu H., Dean M., Li G., Tsang S., Jiang R., Zhang X., Li B., Liu G., Bedekar N., Lu N., Xie G., Liang H., Chang L., Wang T., Chen J., Li Y., Zhang X., Yang H., Xu X., Wang L., Wang J. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. (англ.) // GigaScience. — 2015. — Vol. 4. — P. 37. — doi:10.1186/s13742-015-0068-3. — PMID 26251698.
- Huang L., Ma F., Chapman A., Lu S., Xie X. S. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. (англ.) // Annual review of genomics and human genetics. — 2015. — Vol. 16. — P. 79—102. — doi:10.1146/annurev-genom-090413-025352. — PMID 26077818.
- Chen M., Song P., Zou D., Hu X., Zhao S., Gao S., Ling F. Comparison of multiple displacement amplification (MDA) and multiple annealing and looping-based amplification cycles (MALBAC) in single-cell sequencing. (англ.) // Public Library of Science ONE. — 2014. — Vol. 9, no. 12. — P. e114520. — doi:10.1371/journal.pone.0114520. — PMID 25485707.
- Gawad C., Koh W., Quake S. R. Single-cell genome sequencing: current state of the science. (англ.) // Nature reviews. Genetics. — 2016. — Vol. 17, no. 3. — P. 175—188. — doi:10.1038/nrg.2015.16. — PMID 26806412.
- Abecasis G. R., Auton A., Brooks L. D., DePristo M. A., Durbin R. M., Handsaker R. E., Kang H. M., Marth G. T., McVean G. A. An integrated map of genetic variation from 1,092 human genomes. (англ.) // Nature. — 2012. — Vol. 491, no. 7422. — P. 56—65. — doi:10.1038/nature11632. — PMID 23128226.
- Martin E. R., Lai E. H., Gilbert J. R., Rogala A. R., Afshari A. J., Riley J., Finch K. L., Stevens J. F., Livak K. J., Slotterbeck B. D., Slifer S. H., Warren L. L., Conneally P. M., Schmechel D. E., Purvis I., Pericak-Vance M. A., Roses A. D., Vance J. M. SNPing away at complex diseases: analysis of single-nucleotide polymorphisms around APOE in Alzheimer disease. (англ.) // American journal of human genetics. — 2000. — Vol. 67, no. 2. — P. 383—394. — doi:10.1086/303003. — PMID 10869235.
- Zhu Y., Spitz M. R., Lei L., Mills G. B., Wu X. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter enhances lung cancer susceptibility. (англ.) // Cancer research. — 2001. — Vol. 61, no. 21. — P. 7825—7829. — PMID 11691799.
- Schaschl H., Aitman T. J., Vyse T. J. Copy number variation in the human genome and its implication in autoimmunity. (англ.) // Clinical and experimental immunology. — 2009. — Vol. 156, no. 1. — P. 12—16. — doi:10.1111/j.1365-2249.2008.03865.x. — PMID 19220326.
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. (англ.) // Genome research. — 2010. — Vol. 20, no. 9. — P. 1297—1303. — doi:10.1101/gr.107524.110. — PMID 20644199.
- Zhang J., Wheeler D. A., Yakub I., Wei S., Sood R., Rowe W., Liu P. P., Gibbs R. A., Buetow K. H. SNPdetector: a software tool for sensitive and accurate SNP detection. (англ.) // Public Library of Science for Computational Biology. — 2005. — Vol. 1, no. 5. — P. e53. — doi:10.1371/journal.pcbi.0010053. — PMID 16261194.
- Li R., Li Y., Fang X., Yang H., Wang J., Kristiansen K., Wang J. SNP detection for massively parallel whole-genome resequencing. (англ.) // Genome research. — 2009. — Vol. 19, no. 6. — P. 1124—1132. — doi:10.1101/gr.088013.108. — PMID 19420381.
- Koboldt D. C., Chen K., Wylie T., Larson D. E., McLellan M. D., Mardis E. R., Weinstock G. M., Wilson R. K., Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. (англ.) // Bioinformatics. — 2009. — Vol. 25, no. 17. — P. 2283—2285. — doi:10.1093/bioinformatics/btp373. — PMID 19542151.
- Redon R., Ishikawa S., Fitch K. R., Feuk L., Perry G. H., Andrews T. D., Fiegler H., Shapero M. H., Carson A. R., Chen W., Cho E. K., Dallaire S., Freeman J. L., González J. R., Gratacòs M., Huang J., Kalaitzopoulos D., Komura D., MacDonald J. R., Marshall C. R., Mei R., Montgomery L., Nishimura K., Okamura K., Shen F., Somerville M. J., Tchinda J., Valsesia A., Woodwark C., Yang F., Zhang J., Zerjal T., Zhang J., Armengol L., Conrad D. F., Estivill X., Tyler-Smith C., Carter N. P., Aburatani H., Lee C., Jones K. W., Scherer S. W., Hurles M. E. Global variation in copy number in the human genome. (англ.) // Nature. — 2006. — Vol. 444, no. 7118. — P. 444—454. — doi:10.1038/nature05329. — PMID 17122850.
- Zhang F., Gu W., Hurles M. E., Lupski J. R. Copy number variation in human health, disease, and evolution. (англ.) // Annual review of genomics and human genetics. — 2009. — Vol. 10. — P. 451—481. — doi:10.1146/annurev.genom.9.081307.164217. — PMID 19715442.
- Frank B., Bermejo J. L., Hemminki K., Sutter C., Wappenschmidt B., Meindl A., Kiechle-Bahat M., Bugert P., Schmutzler R. K., Bartram C. R., Burwinkel B. Copy number variant in the candidate tumor suppressor gene MTUS1 and familial breast cancer risk. (англ.) // Carcinogenesis. — 2007. — Vol. 28, no. 7. — P. 1442—1445. — doi:10.1093/carcin/bgm033. — PMID 17301065.
- Glessner J. T., Wang K., Cai G., Korvatska O., Kim C. E., Wood S., Zhang H., Estes A., Brune C. W., Bradfield J. P., Imielinski M., Frackelton E. C., Reichert J., Crawford E. L., Munson J., Sleiman P. M., Chiavacci R., Annaiah K., Thomas K., Hou C., Glaberson W., Flory J., Otieno F., Garris M., Soorya L., Klei L., Piven J., Meyer K. J., Anagnostou E., Sakurai T., Game R. M., Rudd D. S., Zurawiecki D., McDougle C. J., Davis L. K., Miller J., Posey D. J., Michaels S., Kolevzon A., Silverman J. M., Bernier R., Levy S. E., Schultz R. T., Dawson G., Owley T., McMahon W. M., Wassink T. H., Sweeney J. A., Nurnberger J. I., Coon H., Sutcliffe J. S., Minshew N. J., Grant S. F., Bucan M., Cook E. H., Buxbaum J. D., Devlin B., Schellenberg G. D., Hakonarson H. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. (англ.) // Nature. — 2009. — Vol. 459, no. 7246. — P. 569—573. — doi:10.1038/nature07953. — PMID 19404257.
- Xie C., Tammi M. T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. (англ.) // BMC bioinformatics. — 2009. — Vol. 10. — P. 80. — doi:10.1186/1471-2105-10-80. — PMID 19267900.
- Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S. F., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. (англ.) // Genome research. — 2007. — Vol. 17, no. 11. — P. 1665—1674. — doi:10.1101/gr.6861907. — PMID 17921354.
- Ivakhno S., Royce T., Cox A. J., Evers D. J., Cheetham R. K., Tavaré S. CNAseg--a novel framework for identification of copy number changes in cancer from second-generation sequencing data. (англ.) // Bioinformatics. — 2010. — Vol. 26, no. 24. — P. 3051—3058. — doi:10.1093/bioinformatics/btq587. — PMID 20966003.
- Miller C. A., Hampton O., Coarfa C., Milosavljevic A. ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. (англ.) // Public Library of Science ONE. — 2011. — Vol. 6, no. 1. — P. e16327. — doi:10.1371/journal.pone.0016327. — PMID 21305028.
- Klambauer G., Schwarzbauer K., Mayr A., Clevert D. A., Mitterecker A., Bodenhofer U., Hochreiter S. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. (англ.) // Nucleic acids research. — 2012. — Vol. 40, no. 9. — P. e69. — doi:10.1093/nar/gks003. — PMID 22302147.
- Ning L., Liu G., Li G., Hou Y., Tong Y., He J. Current challenges in the bioinformatics of single cell genomics. (англ.) // Frontiers in oncology. — 2014. — Vol. 4. — P. 7. — doi:10.3389/fonc.2014.00007. — PMID 24478987.
- Eisen M. B., Spellman P. T., Brown P. O., Botstein D. Cluster analysis and display of genome-wide expression patterns. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 1998. — Vol. 95, no. 25. — P. 14863—14868. — PMID 9843981.
- Prokopenko D., Hecker J., Silverman E. K., Pagano M., Nöthen M. M., Dina C., Lange C., Fier H. L. Utilizing the Jaccard index to reveal population stratification in sequencing data: a simulation study and an application to the 1000 Genomes Project. (англ.) // Bioinformatics. — 2016. — Vol. 32, no. 9. — P. 1366—1372. — doi:10.1093/bioinformatics/btv752. — PMID 26722118.
- Greaves M., Maley C. C. Clonal evolution in cancer. (англ.) // Nature. — 2012. — Vol. 481, no. 7381. — P. 306—313. — doi:10.1038/nature10762. — PMID 22258609.
- A. P. Dempster; N. M. Laird; D. B. Rubin. Maximum likelihood from incomplete data via the EM algorithm // Journal of the Royal Statistical Society. Series B (Methodological). — 1977. — Vol. 39, № 1. — P. 1—38. — doi:10.2307/2984875.
- C. Fraley, A. E. Raftery. How Many Clusters? Which Clustering Method? Answers Via Model-Based Cluster Analysis (англ.). — 1998. — doi:10.1093/comjnl/41.8.578.
- Chris Fraley, Adrian E. Raftery. MCLUST: Software for Model-Based Cluster Analysis (англ.) // Journal of Classification. — 1999. — doi:10.1007/s003579900058.
- Kim K. I., Simon R. Using single cell sequencing data to model the evolutionary history of a tumor. (англ.) // BMC bioinformatics. — 2014. — Vol. 15. — P. 27. — doi:10.1186/1471-2105-15-27. — PMID 24460695.
- Falconer E., Hills M., Naumann U., Poon S. S., Chavez E. A., Sanders A. D., Zhao Y., Hirst M., Lansdorp P. M. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. (англ.) // Nature methods. — 2012. — Vol. 9, no. 11. — P. 1107—1112. — doi:10.1038/nmeth.2206. — PMID 23042453.
- Falconer E., Lansdorp P. M. Strand-seq: a unifying tool for studies of chromosome segregation. (англ.) // Seminars in cell & developmental biology. — 2013. — Vol. 24, no. 8-9. — P. 643—652. — doi:10.1016/j.semcdb.2013.04.005. — PMID 23665005.
- Coupland P., Chandra T., Quail M., Reik W., Swerdlow H. Direct sequencing of small genomes on the Pacific Biosciences RS without library preparation. (англ.) // BioTechniques. — 2012. — Vol. 53, no. 6. — P. 365—372. — doi:10.2144/000113962. — PMID 23227987.
- El Hajj N., Trapphoff T., Linke M., May A., Hansmann T., Kuhtz J., Reifenberg K., Heinzmann J., Niemann H., Daser A., Eichenlaub-Ritter U., Zechner U., Haaf T. Limiting dilution bisulfite (pyro)sequencing reveals parent-specific methylation patterns in single early mouse embryos and bovine oocytes. (англ.) // Epigenetics. — 2011. — Vol. 6, no. 10. — P. 1176—1188. — doi:10.4161/epi.6.10.17202. — PMID 21937882.
- Guo H., Zhu P., Wu X., Li X., Wen L., Tang F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. (англ.) // Genome research. — 2013. — Vol. 23, no. 12. — P. 2126—2135. — doi:10.1101/gr.161679.113. — PMID 24179143.
- Nagano T., Lubling Y., Yaffe E., Wingett S. W., Dean W., Tanay A., Fraser P. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. (англ.) // Nature protocols. — 2015. — Vol. 10, no. 12. — P. 1986—2003. — doi:10.1038/nprot.2015.127. — PMID 26540590.
- Macaulay I. C., Voet T. Single cell genomics: advances and future perspectives. (англ.) // PLoS genetics. — 2014. — Vol. 10, no. 1. — P. e1004126. — doi:10.1371/journal.pgen.1004126. — PMID 24497842.