Множественное выравнивание последовательностей
Мно́жественное выра́внивание после́довательностей (англ. multiple sequence alignment, MSA) — выравнивание трёх и более биологических последовательностей, обычно белков, ДНК или РНК. В большинстве случаев предполагается, что входной набор последовательностей имеет эволюционную связь. Используя множественное выравнивание, можно оценить эволюционное происхождение последовательностей, проведя филогенетический анализ.

Визуальное представление выравнивания иллюстрирует мутационные события как точечные мутации (изменение одной аминокислоты или одного нуклеотида) в виде различающихся символов в одной колонке выравнивания, а также их вставки и делеции (изображаются знаком дефиса, гэпы).
Множественное выравнивание последовательностей часто используется для оценки консервативности доменов белков, третичных и вторичных структур и даже отдельных аминокислотных остатков или нуклеотидов.
Ввиду большей вычислительной сложности по сравнению с парным выравниванием множественное выравнивание требует более сложные алгоритмы. Многие соответствующие программы используют эвристические алгоритмы, поскольку поиск глобального оптимального выравнивания для многих последовательностей может занимать очень большое время.
Динамическое программирование и вычислительная сложность
Для построения глобального оптимального выравнивания напрямую используется динамическое программирование. Для белковых последовательностей существует два набора параметров: штраф за гэп и матрица замен, содержащая в себе вероятности сопоставления пары аминокислотных остатков, основанных на схожести их химических свойств и эволюционной вероятности мутации. Для нуклеотидных последовательностей также используется штраф за гэп, но матрица замен гораздо проще, в ней учитываются только полные совпадения нуклеотидов или мисматчи, т. е. полные несовпадения[1].
Для n отдельных последовательностей наивный метод требует построения n-мерного эквивалента матрицы, которую используют для парного выравнивания. С ростом n пространство поиска возрастает экспоненциально. Таким образом, наивный алгоритм имеет вычислительную сложность O(Длина последовательностейNпоследовательностей). Поиск глобального оптимума для n последовательностей относится к NP-полным задачам[2][3][4].
В 1989 году на основе алгоритма Каррильо — Липмана[5] Альтшуль представил практический подход, который использовал парные выравнивания для ограничения n-мерного пространства поиска[6]. При таком подходе динамическое программирование выполняется на каждой паре последовательностей из входного набора и ищется только область, расположенная вблизи n-мерного пересечения этих путей. Программа оптимизирует сумму всех пар символов в каждой позиции в выравнивании (сумма парных весов)[7]
Прогрессивное выравнивание
Широко используемый подход — прогрессивное выравнивание, применяющий эвристический алгоритм, разработан Paulien Hogeweg and Ben Hesper в 1984 году[8]. Все методы прогрессивного выравнивания имеют две важные стадии: построение бинарного дерева (путеводное дерево), где листья являются последовательностями, и построение множественного выравнивания путём добавления последовательностей к растущему выравниванию согласно путеводному дереву. Само путеводное дерево может быть построено кластеризующими методами, такими как UPGMA и метод присоединения соседей[9].
Прогрессивное выравнивание не гарантирует получение глобального оптимального выравнивания. Проблема состоит в том, что ошибки, полученные на любой стадии растущего множественного выравнивания, доходят до конечного выравнивания. Кроме того, выравнивание может быть особенно плохим в случае набора сильно отдалённых друг от друга последовательностей. Большинство современных прогрессивных методов имеют изменённую функцию вычисления веса со вторичной весовой функцией, которая присваивает коэффициенты для отдельных элементов набора данных в виде нелинейной моды основанной на их филогенетическом расстоянии от ближайших соседей[9].
Методы прогрессивного выравнивания достаточно эффективны, чтобы применять их для большого числа (100-1000) последовательностей. Самый популярный метод прогрессивного выравнивания принадлежит к семейству Clustal[10], в частности, взвешенный вариант ClustalW[11], доступ к которому можно получить через такие порталы как GenomeNet, EBI, EMBNet Архивная копия от 1 мая 2011 на Wayback Machine. ClustalW активно используется для построения филогенетических деревьев, несмотря на предупреждения автора, что непроверенные вручную выравнивания не должны использоваться ни при построении деревьев, ни в качестве входных данных для предсказания структуры белков. Текущая версия Clustal — Clustal Omega, которая работает на основе путеводных деревьев и HMM профиль-профильных методов для белковых выравниваний. Также предлагаются различные инструменты для построения прогрессивного выравнивания последовательностей ДНК. Один из них – MAFFT (англ. Multiple Alignment using Fast Fourier Transform)[12].
Другой распространённый метод прогрессивного выравнивания, T-Coffee[13], медленнее, чем Clustal и его производные, но в основном даёт более точные выравнивания для отдалённо связанных последовательностей. T-Coffee строит библиотеку парных выравниваний, которую затем использует для построения множественного выравнивания.
Поскольку прогрессивные методы являются эвристическими, они не гарантируют схождения к глобальному оптимуму; качество выравнивания и его биологическое значение бывает трудно оценить. Полупрогрессивный метод, который улучшает качество выравнивания и не использует эвристику c потерями, справляется за полиномиальное время (PSAlign Архивная копия от 18 июля 2011 на Wayback Machine)[14].
Итеративные методы
Набор методов для построения множественных выравниваний, в которых происходит снижение ошибок, наследуемых в прогрессивных методах, классифицируют как «итеративные». Они работают аналогично прогрессивным методам, но при этом неоднократно перестраивают исходные выравнивания при добавлении новых последовательностей. Прогрессивные методы сильно зависят от качества начальных выравниваний, поскольку они в неизменном виде, а стало быть и с ошибками, попадут в конечный результат. Иными словами, если последовательность уже попала в выравнивание, её дальнейшее положение не изменится. Такое приближение улучшает эффективность, но отрицательно сказывается на точности результата. В отличие от прогрессивных методов, итеративные методы могут возвращаться к первоначально посчитанным парным выравниваниям и подвыравниваниям, содержащим подмножества последовательностей из запроса, и таким образом оптимизировать общую целевую функцию и повышать качество[9].
Существует большое количество разнообразных итеративных методов. Например, PRRN/PRRP использует алгоритм восхождения к вершине для оптимизации веса множественного выравнивания[15] и итеративно корректирует веса выравнивания и области со множеством гэпов[9]. PRRP работает эффективнее, когда улучшает выравнивание, предварительно построенное быстрым методом[9].
Ещё одна итеративная программа, DIALIGN, использует необычный подход, сосредотачивая внимание на локальных выравниваниях подсегментов или мотивов последовательностей без введения штрафа за гэп[16]. Выравнивание отдельных мотивов представляется в матричном виде, сходном с диаграммой с точками (dot-plot) в парном выравнивании. Альтернативный метод, который использует быстрые локальные выравнивания как точки заякоривания для более медленной процедуры построения глобального выравнивания, представлен в софте CHAOS/DIALIGN[16].
Третий популярный итерационный метод называется MUSCLE. Он улучшен по сравнению с прогрессивными методами, поскольку использует более точные расстояния для оценки связи двух последовательностей[17]. Расстояния обновляются между итерациями (хотя в первоначальном виде MUSCLE содержал только 2—3 итерации).
Консенсусные методы
Консенсусные методы пытаются выбрать оптимальное множественное выравнивание из различных множественных выравниваний одного и того же набора входных данных. Существуют два наиболее распространённых консенсусных метода: M-COFFEE и MergeAlign[18]. M-COFFEE использует множественные выравнивания, генерируемые 7 различными методами для получения консенсусных выравниваний. MergeAlign способен генерировать консенсусные выравнивания из любого числа входных выравниваний, полученных из различных моделей эволюции последовательности и методов построения. Опция по умолчанию для MergeAlign — выведение консенсусного выравнивания, используя выравнивания, полученные из 91 различных моделей эволюции белковой последовательности.
Скрытые марковские модели
Скрытые марковские модели (HMMs) — вероятностные модели, которые могут оценить правдоподобие для всех возможных комбинаций гэпов, совпадений или несовпадений для того, чтобы определить наиболее вероятное множественное выравнивание или их набор. HMMs могут давать одно выравнивание с высоким весом, но также могут генерировать семейство возможных выравниваний, которые затем могут быть оценены по их биологической значимости. HMMs могут быть использованы для получения как глобальных, так и локальных выравниваний. Несмотря на то, что методы, основанные на HMM, появились сравнительно недавно, они зарекомендовали себя как методы со значительными улучшениями вычислительной сложности, особенно для последовательностей, содержащих перекрывающиеся области[9].
Стандартные методы, основанные на HMM, представляют множественное выравнивание в виде направленного ациклического графа, известного как граф частичного порядка, который состоит из серий узлов, представляющих собой возможные состояния в колонках выравнивания. В этом представлении абсолютно консервативная колонка (т. е. последовательности во множественном выравнивании имеют в этой позиции определённый символ) кодируется как один узел со множеством исходящих соединений с символами, возможными в следующей позиции выравнивания. В терминах стандартной скрытой марковской модели наблюдаемые состояния — отдельные колонки выравнивания, а «скрытые» состояния представляют собой предполагаемую предковую последовательность, от которой последовательности из входного набора могли произойти. Эффективный метод динамического программирования, алгоритм Витерби, широко используется для получения хорошего выравнивания[19]. Он отличается от прогрессивных методов тем, что выравнивание первых последовательностей перестраивается при добавлении каждой новой последовательности. Тем не менее, как и прогрессивные методы, на этот алгоритм может повлиять порядок, в котором последовательности из входного набора поступают в выравнивание, особенно в случае эволюционно слабо связанных последовательностей[9].
Несмотря на то, что HMM-методы более сложны, чем часто используемые прогрессивные методы, существует несколько программ для получения выравниваний, например, POA[20], а также похожий, но более обобщённый метод в пакетах SAM[21] и HMMER[22]. SAM используется для получения выравниваний для предсказания структуры белков в эксперименте CASP для дрожжевых белков. HHsearch, основанный на парном сравнении HMMs, используется для поиска отдалённо связанных последовательностей. Сервер, запускающий HHsearch (HHpred) был самым быстрым из 10 лучших автоматических серверов по предсказанию структур белков в CASP7 и CASP8[23].
Генетические алгоритмы и моделирование отжига
Стандартные оптимизационные методы в компьютерной науке, которые позволяют моделировать, но не прямо воспроизводить физический процесс, также используются для более эффективного построения множественных выравниваний. Один из таких методов, генетический алгоритм, был использован для построения множественного выравнивания последовательностей, основанного на гипотетическом эволюционном процессе, который обеспечил расхождение последовательностей. Этот метод работает с помощью разделения серий возможных MSA на фрагменты и повторной переорганизации этих фрагментов со вводом разрывов в различные позиции. Основная целевая функция оптимизируется в ходе этого процесса обычно с помощью максимизации «сумм пар» методами динамического программирования. Этот метод реализован для белковых последовательностей в программном обеспечении SAGA (англ. Sequence Alignment by Genetic Algorithm)[24], а для последовательностей РНК — в RAGA[25].
С помощью метода моделирования отжига существующее множественное выравнивание, построенное другим методом, уточняется в сериях перестроек для нахождения более хороших участков выравнивания, чем было до этого. Как и в случае генетического алгоритма, при моделировании отжига максимизируется целевая функция как функция сумм пар. В моделировании отжига используется условный «температурный фактор», который определяет уровень протекающих перестроек и уровень правдоподобности каждой перестройки. Типично использование чередующихся периодов с высоким уровнем перестроек и малым уровнем правдоподобия (для обнаружения наиболее удалённых регионов в выравнивании) с периодами с низким уровнем перестроек и высоким уровнем правдоподобия для более тщательного изучения локальных минимумов вблизи новых колонок выравнивания. Этот подход был осуществлён в программе MSASA (англ. Multiple Sequence Alignment by Simulated Annealing)[26].
Методы, основанные на филогенетическом анализе
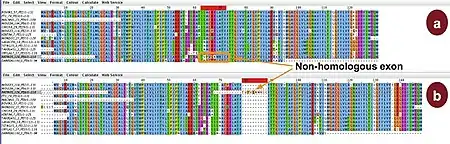
Большинство методов множественного выравнивания старается минимизировать количество вставок/делеций (гэпов), из-за чего продуцирует компактные выравнивания. Такой подход может привести к ошибкам в выравнивании, если выровненные последовательности содержали негомологичные регионы и если гэпы информативны при филогенетическом анализе. Эти проблемы обычны в новых последовательностях, которые бедно аннотированы и могут содержать сдвиги рамки считывания, неправильные домены или негомологичные сплайсированные экзоны.
Первый метод, основанный на анализе филогении, был разработан Лойтиноджом и Голдманом в 2005 году[27]. В 2008 году те же авторы выпустили соответствующее программное обеспечение — PRANK[28]. PRANK совершенствует выравнивания, когда есть вставки. Тем не менее, он работает медленнее, чем прогрессивные и/или итеративные методы[29], которые были разработаны за несколько лет до того.
В 2012 году появились два новых метода, основанных филогенетическом анализе. Первый, названный PAGAN, был разработан командой PRANK, а второй, названный ProGraphMSA, был разработан Жалковским[30]. Их программных обеспечения были разработаны независимо, но имеют общие черты: оба используют алгоритмы на графах для улучшения распознавания негомологичных регионов, а усовершенствования в коде делают их быстрее PRANK.
Поиск мотивов

Поиск мотивов или, иначе, анализ профилей — это метод поиска локализации мотива в глобальном множественном выравнивании как средство получения лучшего MSA и средней величины веса получаемой матрицы с целью использования её для поиска других последовательностей со сходными мотивами. Было разработано множество методов для определения мотивов, но все они основываются на обнаружении коротких высококонсервативных паттернов в большем по длине выравнивания паттерне и конструировании матрицы, подобной матрице замен. Эта матрица отражает нуклеотидный или аминокислотный состав для каждой позиции в предполагаемом мотиве. Затем выравнивание может быть уточнено с помощью этих матриц. В стандартном анализе профилей эта матрица включает в себя вхождения как для каждого возможного символа, так и для гэпа[9]. В противоположность этому, статистический алгоритм поиска паттернов сначала ищет мотивы, а затем использует найденные мотивы для построения множественного выравнивая. Во многих случаях, когда исходное множество последовательностей содержит небольшое количество последовательностей или только высоко родственные последовательности, добавляются псевдокаунты для нормализации распределения, отражённого в весовой матрице. В частности, это помогает избегать нулей в матрице вероятностей, чтобы не получить значение бесконечности в позиционной весовой матрице.
Анализ блоков — это метод поиска мотивов, осуществляемый в регионах выравнивания без гэпов. Блоки могут быть сгенерированы из множественного выравнивания или получены из невыровненных последовательностей путём предварительного расчёта множества общих мотивов из известных семейств генов[31]. Оценка блоков обычно основывается на пространстве символов с высокой частотой встречаемости, а не на вычислении в явном виде матриц замен. Сервер BLOCKS предоставляет альтернативный метод для локализации таких мотивов в невыровненных последовательностях.
Статистическое сопоставление паттернов осуществляется с помощью алгоритма максимизации ожидания и семплирования по Гиббсу. Для поиска мотивов наиболее часто используется сервер MEME, использующий алгоритм максимизации ожидания и метод скрытых марковских моделей, а также MEME/MAST[32][33], использующий дополнительно алгоритм MAST.
Множественное выравнивание некодирующих последовательностей
Некоторые участки ДНК, не кодирующие белок, особенно сайты связывания транскрипционных факторов (TFBS), являются более консервативными и не обязательно эволюционно связанными, так как эти сайты могут встречаться в негомологичных последовательностях. Таким образом, допущения, используемые для выравнивания белковых последовательностей и кодирующих регионов ДНК, не подходят для последовательностей сайтов связывания транскрипционных факторов. Несмотря на то что выравнивание кодирующих белок участков ДНК для гомологичных последовательностей с помощью операторов мутации осмысленно, выравнивание последовательностей сайтов связывания для одного и того же транскрипционного фактора не может основываться на эволюционно связанных мутационных операциях. Аналогичным образом эволюционный оператор точечных мутаций может быть использован для определения редакционного расстояния для кодирующих последовательностей, но малопригоден для последовательностей сайтов связывания транскрипционных факторов из-за того, что любое изменение последовательности должно сохранять определённый уровень специфичности для выполнения функции связывания. Это становится особенно значимо, когда выравнивание последовательностей сайтов связывания транскрипционных факторов нужно с целью построения наблюдаемых моделей для предсказания неизвестных локусов таких же TFBS. Следовательно, методы множественного выравнивания необходимо корректировать, учитывая основные эволюционные гипотезы, и использовать определённые операторы, как в методе EDNA, учитывающем термодинамику для выравнивания сайтов связывания[34].
Визуализация выравнивания и контроль качества
Необходимость использования эвристических подходов для множественного выравнивания приводит к тому, что произвольно выбранное множество белков может с большой вероятностью быть выровнено неверно. Например, оценка некоторых ведущих программ выравнивания при помощи BAliBase benchmark[35] показала, что по меньшей мере 24 % всех выровненных пар аминокислот выровнены неверно[36]. Эти ошибки могут возникать из-за уникальных вставок в один и более участок последовательностей. Они также могут быть обусловлены более сложным эволюционным процессом, приводящим к возникновению белков, которые сложно выровнять только по последовательности, и для качественного выравнивания необходимо знать что-то ещё, например, структуру. По мере увеличения количества выравниваемых последовательностей и их расхождения возрастает ошибка из-за эвристического характера алгоритмов множественного выравнивания. Визуализаторы множественного выравнивания позволяют наглядно оценивать выравнивание часто с помощью проверки качества выравнивания для аннотированных функциональных участков у двух и более последовательностей. Многие визуализаторы также позволяют редактировать выравнивание, корректируя ошибки (обычно минорного характера), для получения оптимального курируемого выравнивания подходящего для использования в филогенетическом анализе или сравнительного моделирования[37].
Как бы то ни было, по мере увеличения числа последовательностей, в особенности в полногеномных исследованиях, которые включают много множественных выравниваний, становится невозможным вручную курировать все выравнивания. Кроме того, ручное курирование субъективно. И, наконец, даже самый лучший специалист не может с уверенностью выровнять многие неоднозначные случаи у сильно разошедшихся последовательностей. В таких случаях обычно практикуется использование автоматических процедур для исключения ненадёжно выровненных регионов множественного выравнивания. С целью получения филогенетических реконструкций широко используется программа Gblocks для удаления блоков выравнивания с предположительно низким качеством, в соответствии с всевозможными границами (катоффами) по количеству последовательностей с гэпами в колонках выравнивания[38]. В то же время эти критерии могут чрезмерно отфильтровывать регионы с вставками/делециями, которые могли бы быть надёжно выровнены, а эти регионы могли бы быть полезны для выявления положительного отбора. Немногие алгоритмы выравнивания выдают сайт-специфичный вес выравнивания, который мог был позволить отбирать высоко консервативные регионы. Такую возможность впервые дала программа SOAP[39], которая тестирует устойчивость каждой колонки к колебанию параметров в популярной программе выравнивания ClustalW. Программа T-Coffee[39] использует библиотеку выравниваний для создания конечного множественного выравнивания и выдаёт множественное выравнивание, окрашенное в соответствии с оценкой доверия, которая отражает соответствие между различными выравниваниями в библиотеке по каждому из выровненных остатков. TCS (англ. Transitive Consistency Score) является её расширением, которое использует библиотеку попарных выравниваний T-Coffee для оценки каждого третьего множественного выравнивания. Попарные проекции могут быть созданы использованием быстрых или медленных методов, таким образом, можно найти компромисс между скоростью и точностью вычислений[40][41]. Другая программа выравнивания, FSA (англ. Fast statistical alignment), использует статистические модели, позволяющие вычислить погрешность выравнивания, и может выдавать множественное выравнивание с оценкой уровня его достоверности. Оценка HoT (англ. Heads-Or-Tails) может быть использована для измерения погрешностей сайт-специфических выравниваний, в которых погрешности могут возникать из-за существования множества ко-оптимальных решений. Программа GUIDANCE[42] вычисляет аналогичную сайт-специфичную меру доверия, базирующуюся на устойчивости выравнивания к неопределённости в направляющем дереве, которое используется, как было сказано выше, в программах прогрессивного выравнивания. В то же время статистически более обоснованным подходом к оценке неопределённостей выравнивания является использование вероятностных эволюционных моделей для совместной оценки филогении и выравнивания. Байесовский подход позволяет рассчитать постериорные вероятности оценок филогении и выравнивания, которые измеряют уровень уверенности в этих оценках. В этом случае постерионная вероятность может быть вычислена для каждого сайта в выравнивании. Такой подход реализован в программе Bali-Phy[43].
Использование в филогенетике
Множественное выравнивание последовательностей может быть использовано для построения филогенетического дерева[44]. Это возможно по двум причинам. Во-первых, функциональные домены, известные для аннотированных последовательностей, могут быть использованы для выравнивания неаннотированных последовательностей. Во-вторых, консервативные регионы могут иметь функциональную значимость. Из-за этого возможно использование множественных выравниваний для анализа и нахождения эволюционных связей через гомологию последовательностей. Точечные мутации и вставки/деления могут быть также обнаружены[45].
Определение местоположения консервативных доменов с помощью множественного выравнивания может быть также использовано для идентифицированная функционально важных сайтов, таких как сайты связывания, регуляторные сайты или сайты ответственные за другие ключевые функции. При анализе множественного выравнивания полезно рассматривать различные характеристики. К таким полезным характеристикам выравнивания относятся идентичность, схожесть и гомология последовательностей. Идентичность определяет, что последовательности имеют одинаковые остатки в соответствующих положениях. Схожесть определяется сходными остатками в количественном соотношении. Например, с точки зрения нуклеотидных последовательностей пиримидины считаются похожими между собой, как и пурины. Сходство в конечном счёте приводит к гомологии, так, чем более сходны последовательности, тем более близкими гомологами они являются. Также сходство последовательностей может помочь в нахождении общего происхождения[46].
Примечания
- Help with matrices used in sequence comparison tools (недоступная ссылка). European Bioinformatics Institute. Дата обращения: 3 марта 2010. Архивировано 11 марта 2010 года.
- Wang L., Jiang T. On the complexity of multiple sequence alignment. (англ.) // Journal of computational biology : a journal of computational molecular cell biology. — 1994. — Vol. 1, no. 4. — P. 337—348. — doi:10.1089/cmb.1994.1.337. — PMID 8790475.
- Just W. Computational complexity of multiple sequence alignment with SP-score. (англ.) // Journal of computational biology : a journal of computational molecular cell biology. — 2001. — Vol. 8, no. 6. — P. 615—623. — doi:10.1089/106652701753307511. — PMID 11747615.
- Elias I. Settling the intractability of multiple alignment. (англ.) // Journal of computational biology : a journal of computational molecular cell biology. — 2006. — Vol. 13, no. 7. — P. 1323—1339. — doi:10.1089/cmb.2006.13.1323. — PMID 17037961.
- Carrillo H., Lipman D. J. The Multiple Sequence Alignment Problem in Biology (англ.) // SIAM Journal of Applied Mathematics : journal. — 1988. — Vol. 48, no. 5. — P. 1073—1082. — doi:10.1137/0148063.
- Lipman D. J., Altschul S. F., Kececioglu J. D. A tool for multiple sequence alignment. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 1989. — Vol. 86, no. 12. — P. 4412—4415. — PMID 2734293.
- Genetic analysis software. National Center for Biotechnology Information. Дата обращения: 3 марта 2010.
- Hogeweg P., Hesper B. The alignment of sets of sequences and the construction of phyletic trees: an integrated method. (англ.) // Journal of molecular evolution. — 1984. — Vol. 20, no. 2. — P. 175—186. — PMID 6433036.
- Mount D. M. Bioinformatics: Sequence and Genome Analysis 2nd ed. (англ.) // Cold Spring Harbor : journal. — 2004.
- Higgins D. G., Sharp P. M. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. (англ.) // Gene. — 1988. — Vol. 73, no. 1. — P. 237—244. — PMID 3243435.
- Thompson J. D., Higgins D. G., Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. (англ.) // Nucleic acids research. — 1994. — Vol. 22, no. 22. — P. 4673—4680. — PMID 7984417.
- EMBL-EBI-ClustalW2-Multiple Sequence Alignment. CLUSTALW2.
- Notredame C., Higgins D. G., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. (англ.) // Journal of molecular biology. — 2000. — Vol. 302, no. 1. — P. 205—217. — doi:10.1006/jmbi.2000.4042. — PMID 10964570.
- Sze S. H., Lu Y., Yang Q. A polynomial time solvable formulation of multiple sequence alignment. (англ.) // Journal of computational biology : a journal of computational molecular cell biology. — 2006. — Vol. 13, no. 2. — P. 309—319. — doi:10.1089/cmb.2006.13.309. — PMID 16597242.
- Gotoh O. Significant improvement in accuracy of multiple protein sequence alignments by iterative refinement as assessed by reference to structural alignments. (англ.) // Journal of molecular biology. — 1996. — Vol. 264, no. 4. — P. 823—838. — doi:10.1006/jmbi.1996.0679. — PMID 8980688.
- Brudno M., Chapman M., Göttgens B., Batzoglou S., Morgenstern B. Fast and sensitive multiple alignment of large genomic sequences. (англ.) // BMC bioinformatics. — 2003. — Vol. 4. — P. 66. — doi:10.1186/1471-2105-4-66. — PMID 14693042.
- Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. (англ.) // Nucleic acids research. — 2004. — Vol. 32, no. 5. — P. 1792—1797. — doi:10.1093/nar/gkh340. — PMID 15034147.
- Collingridge P. W., Kelly S. MergeAlign: improving multiple sequence alignment performance by dynamic reconstruction of consensus multiple sequence alignments. (англ.) // BMC bioinformatics. — 2012. — Vol. 13. — P. 117. — doi:10.1186/1471-2105-13-117. — PMID 22646090.
- Hughey R., Krogh A. Hidden Markov models for sequence analysis: extension and analysis of the basic method. (англ.) // Computer applications in the biosciences : CABIOS. — 1996. — Vol. 12, no. 2. — P. 95—107. — PMID 8744772.
- Grasso C., Lee C. Combining partial order alignment and progressive multiple sequence alignment increases alignment speed and scalability to very large alignment problems. (англ.) // Bioinformatics. — 2004. — Vol. 20, no. 10. — P. 1546—1556. — doi:10.1093/bioinformatics/bth126. — PMID 14962922.
- Hughey R, Krogh A. SAM: Sequence alignment and modeling software system. Technical Report UCSC-CRL-96-22, University of California, Santa Cruz, CA, September 1996.
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological sequence analysis: probabilistic models of proteins and nucleic acids. — Cambridge University Press, 1998. — ISBN 0-521-63041-4.
- Battey J. N., Kopp J., Bordoli L., Read R. J., Clarke N. D., Schwede T. Automated server predictions in CASP7. (англ.) // Proteins. — 2007. — Vol. 69 Suppl 8. — P. 68—82. — doi:10.1002/prot.21761. — PMID 17894354.
- Notredame C., Higgins D. G. SAGA: sequence alignment by genetic algorithm. (англ.) // Nucleic acids research. — 1996. — Vol. 24, no. 8. — P. 1515—1524. — PMID 8628686.
- Notredame C., O'Brien E. A., Higgins D. G. RAGA: RNA sequence alignment by genetic algorithm. (англ.) // Nucleic acids research. — 1997. — Vol. 25, no. 22. — P. 4570—4580. — PMID 9358168.
- Kim J., Pramanik S., Chung M. J. Multiple sequence alignment using simulated annealing. (англ.) // Computer applications in the biosciences : CABIOS. — 1994. — Vol. 10, no. 4. — P. 419—426. — PMID 7804875.
- Löytynoja A., Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2005. — Vol. 102, no. 30. — P. 10557—10562. — doi:10.1073/pnas.0409137102. — PMID 16000407.
- Löytynoja A., Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. (англ.) // Science (New York, N.Y.). — 2008. — Vol. 320, no. 5883. — P. 1632—1635. — doi:10.1126/science.1158395. — PMID 18566285.
- Lupyan D., Leo-Macias A., Ortiz A. R. A new progressive-iterative algorithm for multiple structure alignment. (англ.) // Bioinformatics. — 2005. — Vol. 21, no. 15. — P. 3255—3263. — doi:10.1093/bioinformatics/bti527. — PMID 15941743.
- Szalkowski A. M. Fast and robust multiple sequence alignment with phylogeny-aware gap placement. (англ.) // BMC bioinformatics. — 2012. — Vol. 13. — P. 129. — doi:10.1186/1471-2105-13-129. — PMID 22694311.
- Henikoff S., Henikoff J. G. Automated assembly of protein blocks for database searching. (англ.) // Nucleic acids research. — 1991. — Vol. 19, no. 23. — P. 6565—6572. — PMID 1754394.
- Bailey T. L., Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. (англ.) // Proceedings / ... International Conference on Intelligent Systems for Molecular Biology ; ISMB. International Conference on Intelligent Systems for Molecular Biology. — 1994. — Vol. 2. — P. 28—36. — PMID 7584402.
- Bailey T. L., Gribskov M. Combining evidence using p-values: application to sequence homology searches. (англ.) // Bioinformatics. — 1998. — Vol. 14, no. 1. — P. 48—54. — PMID 9520501.
- Salama R. A., Stekel D. J. A non-independent energy-based multiple sequence alignment improves prediction of transcription factor binding sites. (англ.) // Bioinformatics. — 2013. — Vol. 29, no. 21. — P. 2699—2704. — doi:10.1093/bioinformatics/btt463. — PMID 23990411.
- Bahr A., Thompson J. D., Thierry J. C., Poch O. BAliBASE (Benchmark Alignment dataBASE): enhancements for repeats, transmembrane sequences and circular permutations. (англ.) // Nucleic acids research. — 2001. — Vol. 29, no. 1. — P. 323—326. — PMID 11125126.
- Nuin P. A., Wang Z., Tillier E. R. The accuracy of several multiple sequence alignment programs for proteins. (англ.) // BMC bioinformatics. — 2006. — Vol. 7. — P. 471. — doi:10.1186/1471-2105-7-471. — PMID 17062146.
- Aidan Budd. Manual Editing and Adjustment of MSAs (Multiple Sequence Alignments) (недоступная ссылка). www.embl.de. Дата обращения: 23 апреля 2016. Архивировано 24 сентября 2015 года.
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. (англ.) // Molecular biology and evolution. — 2000. — Vol. 17, no. 4. — P. 540—552. — PMID 10742046.
- Löytynoja A., Milinkovitch M. C. SOAP, cleaning multiple alignments from unstable blocks. (англ.) // Bioinformatics. — 2001. — Vol. 17, no. 6. — P. 573—574. — PMID 11395440.
- Chang J. M., Di Tommaso P., Notredame C. TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction. (англ.) // Molecular biology and evolution. — 2014. — Vol. 31, no. 6. — P. 1625—1637. — doi:10.1093/molbev/msu117. — PMID 24694831.
- Chang J. M., Di Tommaso P., Lefort V., Gascuel O., Notredame C. TCS: a web server for multiple sequence alignment evaluation and phylogenetic reconstruction. (англ.) // Nucleic acids research. — 2015. — Vol. 43, no. W1. — P. 3—6. — doi:10.1093/nar/gkv310. — PMID 25855806.
- Penn O., Privman E., Landan G., Graur D., Pupko T. An alignment confidence score capturing robustness to guide tree uncertainty. (англ.) // Molecular biology and evolution. — 2010. — Vol. 27, no. 8. — P. 1759—1767. — doi:10.1093/molbev/msq066. — PMID 20207713.
- Redelings B. D., Suchard M. A. Joint Bayesian estimation of alignment and phylogeny. (англ.) // Systematic biology. — 2005. — Vol. 54, no. 3. — P. 401—418. — doi:10.1080/10635150590947041. — PMID 16012107.
- Kumar, S., and Filipski, A. Multiple sequence alignment: in pursuit of homologous DNA positions // Genome research. — 2007. — Vol. 17, № 2. — P. 127-135. — doi:10.1101/gr.5232407.
- Barton, N. H., Briggs, D. E. G., Eisen, J. A., Goldstein, D. B., and Patel, N. H. Phylogenetic Reconstruction // Evolution. — Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press, 2007. — ISBN 978-0-87969-684-9.
T. A. Brown. The Reconstruction of DNA-based Phylogenetic Trees // Genomes 3. — Garland Science, 2007. — P. 599-609. — ISBN 0-8153-4138-5. - Aidan Budd. Multiple Sequence Alignments: Exercises and Demonstrations (недоступная ссылка). www.embl.de. Дата обращения: 23 апреля 2016. Архивировано 5 марта 2012 года.