Спектроскопия в ближней инфракрасной области
Спектроскопия в ближней инфракрасной области (БИК-спектроскопия, англ. near-infrared spectroscopy, NIR) — раздел спектроскопии, изучающий взаимодействие ближнего инфракрасного излучения (от 780 до 2500 нм, или от 12 800 до 4000 см-1) с веществами. Область ближнего инфракрасного излучения располагается между видимым светом и средней инфракрасной областью.
История

Инфракрасное излучение было открыто в 1800 году английским астрономом Уильямом Гершелем. Разложив видимый свет в спектр при помощи большой стеклянной призмы, он заметил, что нагревание термометра увеличивается от фиолетовой части спектра к красной, а максимальный эффект наблюдается уже за пределами видимого спектра. Дальнейший прогресс произошёл в 1880-х годах, когда Уильям Эбни и Эдвард Фестинг записали несколько спектров органических жидкостей в области от 1 до 1,2 мкм. Американский физик Уильям Кобленц сконструировал собственный спектрометр, использующий в качестве монохроматора призму из каменной соли. Несмотря на то, что прибор был очень чувствителен к внешним колебаниям, а на снятие одного спектра уходил целый день, Кобленц смог записать спектры нескольких сотен веществ в области длин волн от 1 до 15 мкм и даже заметил некоторые спектральные сходства у определённых классов соединений[1].
Между открытием и рутинным использованием инфракрасной спектроскопии прошло около полувека: лишь немногие учёные имели доступ к ИК-спектрометрам. К тому же их устройство было далёким от совершенного. Что касается спектроскопии в ближней инфракрасной области, то ей потребовалось ещё больше времени. Она начала применяться лишь примерно через 70 лет после открытия. К 1970 году в печати появилось лишь 70 статей, касающихся этой разновидности спектроскопии. В 1930-е годы в связи с приближением Второй мировой войны происходило изучение сульфида свинца как детектора тепла, и уже в 1950-е годы он появился на рынке как очень чувствительный детектор излучения в области от 1 до 2,5 мкм[1].
Исследование спектров в ближней ИК-области, по сравнению с инфракрасными спектрами, проводилось очень медленными темпами. Спектроскописты считали эту область слишком сложной для интерпретации. Кроме того, сигналы имели очень небольшую интенсивность (на 2-3 порядка ниже, чем сигналы в ИК-области), а базовую линию трудно было определить из-за большого числа сигналов. Однако метод имел и достоинства. Во-первых, это весьма чувствительный детектор из сульфида свинца и возможность использования вольфрамовых ламп накаливания в качестве источников излучения. Благодаря этим компонентам была возможность снимать спектры диффузного отражения. Во-вторых, можно было создать дешёвые спектрометры, поскольку источники, детекторы и оптика из обычного стекла были достаточно доступны[1].
В 1950-х годах появилась большая потребность в количественном определении воды, белков и жиров. Для этих целей сотрудники министерства сельского хозяйства США избрали БИК-спектроскопию и уже в 1970 годах разработали спектрометры и методики их калибровки для сельскохозяйственных нужд. В 1980-е годы спектрометры стали управляться компьютерами[1].
Принцип метода
Приближение гармонического осциллятора
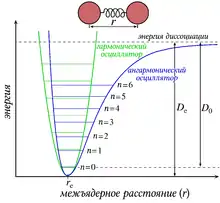
Колебания двухатомной молекулы можно приближенно описать при помощи модели гармонического осциллятора. Согласно этой модели, двухатомная молекула рассматривается как две точечные массы m1 и m2, соединённые упругой невесомой пружиной с коэффициентом упругости (силовой постоянной) K. Потенциальная энергия V такой системы связана с отклонением масс q, возникающим в результате колебаний[2].

Вследствие этого, кривая потенциальной энергии имеет параболическую форму и симметрична относительно равновесной длины колеблющейся связи. Частота колебаний пружины также связана с силовой постоянной и приведённой массой μ[2].

Если частоту колебаний выражать в волновых числах, то это выражение примет следующий вид[2]:

Квантовомеханическое решение уравнения Шрёдингера показывает, что энергия колебаний квантована, т. е. принимает определённые дискретные значения, согласно уравнению[2]:

где h — постоянная Планка, а n — колебательное квантовое число, принимающее целые значения 0, 1, 2, 3... и т. д. Таким образом, колебательные уровни находятся на одинаковом расстоянии друг от друга. Согласно распределению Больцмана, большинство молекул при комнатной температуре занимает нулевой колебательный уровень. Поскольку в инфракрасной спектроскопии правилами отбора разрешены переходы только на соседний колебательный уровень, так что[2]:

в ИК-спектрах наблюдаются, в основном, лишь переходы с нулевого на первый колебательный уровень, так называемые фундаментальные переходы. Большинство из них попадает в область от 4000 до 200 см–1.[2]
Приближение ангармонического осциллятора
Модель гармонического осциллятора не вполне точно описывает колебания молекул, поскольку она не учитывает того, что между атомами при одной связи существуют силы отталкивания и что при сильном удлинении связи может произойти диссоциация. Согласно более точной модели ангармонического осциллятора, энергия колебательных уровней выражается следующим образом[3]:

где χ — постоянная ангармоничности. Таким образом, кривая потенциальной энергии приобретает несимметричную форму (кривая Морзе). Расстояние между уровнями больше не является одинаковым, и правило отбора, запрещающее переходы на дальние колебательные уровни не выполняется так строго. В связи с этим, к числу возможных фундаментальных колебаний молекулы, которое равно 3n–6(5), добавляются ранее запрещённые переходы — обертоны, соответствующие[3]

Кроме обертонов, в инфракрасной спектроскопии могут также наблюдаться составные (комбинационные) полосы, которые представляют собой сумму либо разность фундаментальных колебаний. Именно эти два типа полос и проявляются в ближней инфракрасной области. Однако и обертоны, и составные полосы имеют гораздо меньшую интенсивность, чем фундаментальные полосы, а их наложение друг на друга усложняет интерпретацию БИК-спектров[3].
Расчёт волновых чисел обертонов
| Колебание | Константа χ[4] |
|---|---|
| ν(С–Н) | ~1,9·10–2 |
| ν(С–D) | ~1,5·10–2 |
| ν(С–F) | ~4·10–3 |
| ν(С–Cl) | ~6·10–3 |
| ν(С=O) | ~6,5·10–3 |
Энергия фундаментального колебания или обертона, а также значение волнового числа, при котором они проявляются, могут быть вычислены следующим образом:


Однако поскольку волновое число ṽ0 не может быть напрямую получено из спектральных данных, его необходимо выразить через волновое число ṽ1 и подставить в выражение для волнового числа обертона:


Полученное выражение позволяет найти волновое число для любого обертона с n = 2, 3, 4..., исходя из волнового числа фундаментального колебания, а также константы ангармоничности χ. Также по нему можно рассчитать и константу χ, если известны, например, ṽ1 и ṽ2[4].
Интенсивности обертонов также связаны с ангармоничностью. Показано, что колебания с низкими константами ангармоничности дают обертоны с низкими интенсивностями. Напротив, валентные колебания Х–Н, обладающие высокой ангармоничностью, доминируют в спектрах ближней инфракрасной области[4].
Сравнение со смежными методами
| Переход | Положение полосы | ε, см²/моль | |
|---|---|---|---|
| нм | см–1 | ||
| фундаментальный | 3290 | 3040 | 25 000 |
| обертон 1 | 1693 | 5907 | 1620 |
| обертон 2 | 1154 | 8666 | 48 |
| обертон 3 | 882 | 11 338 | 1,7 |
| обертон 4 | 724 | 13 831 | 0,15 |
Спектроскопия в ближней инфракрасной области основана на использовании излучения в спектральной области от 12 800 до 4000 см—1. В эту область попадают, в основном, обертоны и составные полосы. Поскольку вероятность таких переходов сравнительно мала, интенсивность соответствующих полос понижается примерно на 1-2 порядка на каждый шаг от фундаментального колебания. То есть коэффициент экстинкции ε для обертона с Δn = 3 примерно в 100 раз ниже, чем для обертона с Δn = 2. Таким образом, при движении от ИК-области в сторону области видимого излучения интенсивность полос сильно уменьшается, поскольку ближе к области видимого излучения наблюдаются переходы с большими Δn, а вероятность таких переходов мала. В этом состоит существенное отличие БИК-спектроскопии от ИК-спектроскопии и КР-спектроскопии, где сигналы во всей спектральной области произвольно меняют интенсивность в зависимости от условий возбуждения каждого конкретного колебания[6][5].
В отличие от ИК- и КР-спектроскопии, БИК-спектроскопия требует наличия большой ангармоничности колебаний, поэтому в ближней инфракрасной области проявляются, в основном, колебания С–Н, N–H и O–H. Этому феномену способствует также то, что фундаментальные колебания этих групп проявляются при волновых числах выше 2000 см–1, поэтому их обертоны находятся уже в БИК-области. С другой стороны, наиболее распространённые и интенсивные колебания в ИК-спектрах проявляются при волновых числах ниже 2000 см–1, поэтому их первые обертоны находятся всё ещё за пределами ближней инфракрасной области. Поскольку же интенсивность второго и третьего обертонов резко падает, они практически не видны в спектрах. Например, интенсивное колебание группы C–F происходит при 1600 см–1, однако из-за низкой константы ангармоничности первый и второй обертон находятся при 2400 и 3600 см–1 и не попадают в БИК-область[6].
Наложение многих обертонов и составных сигналов в ближней инфракрасной области приводит к уменьшению структурной селективности БИК-спектров, в сравнении с ИК- или КР-спектрами, в которых фундаментальные колебания обычно чётко отделены друг от друга. Тем не менее, сигналы могут быть соотнесены с соответствующими колебаниями либо путём расчёта частоты обертонов, либо при помощи хемометрических методов[6].
Как и ИК-спектроскопия, БИК-спектроскопия подчиняется закону Бугера — Ламберта — Бера, то есть интенсивность пропускаемого света связана с концентрацией раствора и длиной оптического пути через кювету. Напротив, интенсивность излучения в КР-спектроскопии определяется лишь концентрацией определяемого вещества[6].
| Характеристики[7] | КР-спектроскопия | ИК-спектроскопия | БИК-спектроскопия |
|---|---|---|---|
| тип спектроскопии | спектроскопия рассеяния | спектроскопия поглощения | |
| переходы | |||
| полосы | фундаментальные 4000—50 см-1 |
фундаментальные 4000—200 см-1 |
обертоны и комбинационные 12 500—4000 см-1 |
| излучение | монохроматическое | полихроматическое | |
| правила отбора | изменение поляризуемости |
изменение дипольного момента |
ангармоничность |
| наблюдаемые группы | гомоядерные (например, C=C) |
полярные (например, C=O) |
группы CH, OH, NH |
| структурная селективность | высокая | низкая | |
| количественная характеристика | |||





Примечания
- Burns, Ciurczak, 2007, p. 3—6.
- Burns, Ciurczak, 2007, p. 9—11.
- Burns, Ciurczak, 2007, p. 11—12.
- Burns, Ciurczak, 2007, p. 12.
- Бёккер, 2009, p. 196–199.
- Burns, Ciurczak, 2007, p. 15—16.
- Burns, Ciurczak, 2007, p. 9, 11.
Литература
- Бёккер Ю. Спектроскопия = Spektroskopie / Пер. с нем. Л. Н. Казанцевой, под ред. А. А. Пупышева, М. В. Поляковой. — М.: Техносфера, 2009. — 528 с. — ISBN 978-5-94836-220-5.
- Encyclopedia of Spectroscopy and Spectrometry / Lindon J. — 2nd Ed. — Academic Press, 2010. — 3312 p.
- Boas, D. A. Haemoglobin oxygen saturation as a biomarker: the problem and a solution : [англ.] / D. A. Boas, M. A. Franceschini // Philosophical Transactions of the Royal Society A. — 2011. — Vol. 369, no. 1955. — P. 4407-4424. — doi:10.1098/rsta.2011.0250. — PMID 22006898.
- Burns D. A., Ciurczak E. W. Handbook of Near-Infrared Analysis. — CRC Press, 2007. — 826 p. — ISBN 978-0-8493-7393-0.
- Scheeren, T. W. L. Monitoring tissue oxygenation by near infrared spectroscopy (NIRS): background and current applications : [англ.] / T. W. L. Scheeren, P. Schober, L. A. Schwarte // Journal of Clinical Monitoring and Computing. — 2012. — Vol. 26, no. 4. — P. 279-287. — doi:10.1007/s10877-012-9348-y. — PMID 22467064.