STRING
STRING (сокр. от англ. Search Tool for the Retrieval of Interacting Genes/Proteins) — база данных и веб-ресурс для поиска информации об известных и предсказанных белок-белковых взаимодействиях[1][2][3][4][5][6][7][8].
| STRING | |
|---|---|
 | |
| Содержимое | |
| Описание | Биоинформатический ресурс об известных и предсказанных белок-белковых взаимодействиях |
| Организмы | Все |
| Контакты | |
| Лаборатория | CPR, EMBL, KU, SIB, TUD, UZH |
| Дата выпуска | 2000 |
| Доступность | |
| Сайт | STRING |
| Прочее | |
| Версия | 10.5 (2017) |
STRING обобщает информацию из различных источников: экспериментальные данные, литературные данные и предсказания de novo. Версия 10 содержит информацию о взаимодействиях 9 643 763 белков в 2031 виде организмов, от бактерий и архей до человека. База данных регулярно обновляется и доступна для свободного скачивания[1].
STRING разработан консорциумом европейских университетов CPR, EMBL, KU, SIB, TUD и UZH.
Источники данных
В STRING основная единица — функциональная взаимосвязь, т.е. специфичная и биологически значимая функциональная связь между двумя белками [3].
Для каждой функциональной взаимосвязи STRING рассчитывает оценку достоверности, интегрирующую различные типы доказательств данной взаимосвязи (экспериментальные данные, литературные данные и предсказания de novo на основании ортологии экспериментально изученным белкам, а также на основании сравнительного анализа геномного контекста [9]). Такой комплексный подход имеет следующие преимущества [6]:
- На один стабильный набор белков отображаются различные типы доказательств, облегчая сравнительный анализ.
- Известные и предсказанные взаимодействия зачастую частично дополняют друг друга, что ведёт к расширению сети взаимодействий (на заданном уровне достоверности).
- Оценка достоверности функциональной взаимосвязи повышается, когда данная взаимосвязь подтверждается несколькими типами доказательств.
- Предсказание взаимодействий для большого числа организмов облегчает эволюционный анализ.
При расчёте оценки достоверности функциональной взаимосвязи различные типы доказательств данной взаимосвязи считаются независимыми и оценка рассчитывается по следующей формуле [6]:
где — вклад одного типа доказательств.


STRING не содержит информации о механизме белок-белковых взаимодействий, а также о том, в какое время клеточного цикла может иметь место данное взаимодействие, как оно зависит от внешних условий и насколько оно тканеспецифично. Напротив, STRING содержит информацию о всех возможных белок-белковых взаимодействиях в данном организме, в том числе информацию, предсказанную с определённой достоверностью, что делает STRING наиболее полным ресурсом о белок-белковых взаимодействиях, доступным на сегодняшний день, и особенно полезным для поиска информации о белках, не изученных экспериментально[4].
Экспериментальные данные
STRING интегрирует информацию о взаимодействиях белков в структурных комплексах и метаболических путях, заимствованную из баз данных BIND, BioCarta, BioCyc, BioGRID, DIP, DISEASES, GO, HPRD, IntAct, KEGG, MINT, NCI-Nature Pathway Interaction Database, PDB, Reactome, TISSUES[1][3].
Литературные данные
STRING извлекает информацию о взаимодействиях белков из полных текстов статей из баз данных PubMed, SGD, OMIM, FLyBase и из аннотаций статей из базы данных MEDLINE. Для этого в текстах производится автоматический поиск статистически значимых совместных упоминаний названий генов и их синонимов (данные о синонимах берутся из Swiss-Prot) с использованием обработки естественного языка. Для увеличения точности разработана оценочная система, учитывающая совместное упоминание названий генов в предложениях, абзацах и полных текстах статей[2].
Предсказания de novo
STRING стремится дополнить функциональную аннотацию вновь секвенированных геномов путём de novo предсказаний функциональных взаимосвязей на основании ортологии экспериментально изученным белкам, а также на основании сравнительного анализа геномного контекста[9]. STRING также даёт собственную оценку экспериментально изученным функциональным взаимосвязям, дополняя информацию о них.
Импорт полностью секвенированных геномов
Начиная с версии 9 (2011), STRING импортирует для анализа полностью секвенированные геномы, доступные в базах данных RefSeq и Ensembl, а также на специализированных сайтах[3]. Импортированные геномы предварительно проверяются вручную на предмет полноты и неизбыточности. STRING не хранит информацию о различных изоформах белка, полученных в результате альтернативного сплайсинга или посттрансляционной модификации. Напротив, STRING ставит в соответствие одному локусу одну изоформу белка (как правило, наиболее длинную изоформу)[5]. Такая фильтрация необходима для нормальной работы алгоритмов предсказания белок-белковых взаимодействий.
Предсказания взаимодействий белков на основании ортологии с экспериментально изученными белками
STRING считает референсным взаимодействие белков, участвующих в одном метаболическом пути KEGG, поскольку эта база данных курируется вручную и охватывает ряд организмов и функциональных областей. STRING переносит взаимодействия белков, описанные в метаболических путях KEGG, на ортологичные белки других организмов и присваивает каждому предсказанному белок-белковому взаимодействию определённый вес, который соответствует вероятности нахождения данных белков в одном метаболическом пути KEGG[6] и вносит вклад в итоговую оценку достоверности данной функциональной взаимосвязи.
До версии 8 (2009) предсказания на основании ортологии с белками, описанными в метаболических путях KEGG, производились с использованием кластеров ортологичных групп белков (COGs)[10], затем стали использоваться иерархические ортологичные группы белков из базы данных eggNOG [11].
Начиная с версии 9.1 (2013) предсказания на основании ортологии с белками, описанными в метаболических путях KEGG, производятся с учётом таксономии организмов, что позволяет избежать ошибочного переноса взаимодействия белков одного организма на предполагаемые ортологичные белки другого организма при наличии паралогов данных белков в другом организме, которые возникли вследствие дупликации соответствующих генов в процессе эволюции. Используется версия таксономии, поддерживаемая NCBI. Перенос белок-белковых взаимодействий между организмами на основании ортологии производится последовательно от низших к высшим уровням таксономической иерархии[2].
Предсказания на основании сравнительного анализа геномного контекста
Гены, белковые продукты которых совместно функционируют в метаболическом пути или структурном комплексе, часто имеют общую регуляцию и испытывают общее давление естественного отбора. Такие гены имеют тенденцию к ко-локализации[12] и даже к образованию фьюжн-гена[13]. Часто такие гены находятся близко друг к другу, предположительно являясь одной транскрипционной единицей (опероном). В оперонах разных организмов набор генов и их порядок похожи, но не обязательно идентичны. STRING различает следующие типы геномного контекста[9]:
- Фьюжн-ген, кодирующий фьюжн-белок.
- Консервативное окружение гена (характерно для близкородственных прокариот).
- Совместно встречающиеся гены (характерно для прокариот).
- Совместно экспрессирующиеся гены.
У эукариот не наблюдается оперонных структур, но некоторые эукариотические белки ортологичны прокариотическим белкам, поэтому STRING переносит на эукариотические белки функциональные взаимосвязи, предсказанные на основании сравнительного анализа геномного контекста у прокариот[8].
STRING производит поиск консервативных генных кластеров, эволюционные истории которых похожи сильнее, чем ожидалось бы случайно. STRING стартует с одного гена-затравки и на первой итерации находит гены, которые часто встречаются с данным геном в одном геномном контексте у многих филогенетически далёких организмов. Идеального совпадения между встречаемостью генов не требуется, хотя эта информация оценивается количественно. На следующей итерации в качестве затравок используются новые гены, найденные на предыдущей итерации. Итерации продолжаются до тех пор, пока не будет найдено ни одного нового гена (сходимость). Таким образом, находится множество генов, косвенно связанных с геном-затравкой. Допускается вхождение в один геномный контекст только генов, расстояния между которыми не более 300 пар нуклеотидов[8]. Начиная с версии 8 допускается вхождение в один геномный контекст генов, расположенных на разных цепях ДНК. В последнем случае предсказанной функциональной взаимосвязи присваивается меньший вес, вносящий меньший вклад в итоговую оценку достоверности данной взаимосвязи, по сравнению с функциональной взаимосвязью, предсказанной по геномному контексту, состоящему из генов, расположенных только на одной цепи ДНК[4]. Присваеваемый вес нормируется на число организмов, у которых предсказана данная взаимосвязь[7], и увеличивается при предсказании данной взаимосвязи у филогенетически далёких орнанизмов[6].
При сборке консервативного окружения гена начиная с версии 8 игнорируются короткие частично перекрывающиеся гены на некодирующей цепи ДНК, т.к. они могут оказаться ложными предсказаниями[4].
Начиная с 2005 года в STRING имеются два подхода к предсказанию белок-белковых взаимодействий на основании сравнительного анализа геномного контекста: при запросе пользователь может выбрать COGs-режим или Proteins-режим. В COGs-режиме поиск консервативных генных кластеров производится с требованием ортологичности белков, т.е. взаимодействия предсказываются по принципу «всё или ничего». В Proteins-режиме поиск консервативных генных кластеров производится по количественному сходству аминокислотных последовательностей белков, т.е. предсказываемые взаимодействия могут быть распространены на паралоги, если они есть в организме[6]. Ранее в STRING количественное сходство аминокислотных последовательностей белков определялось по алгоритму Смита — Ватермана. Начиная с версии 9 (2011) для количественного определения сходства аминокислотных последовательностей белков используются матрицы SIMAP[3][14].
Пользовательский интерфейс
Для того, чтобы сделать запрос в базу данных STRING, нужно указать идентификатор или аминокислотную последовательность одного или нескольких белков, а также выбрать организм. В случае запроса для аминокислотной последовательности белка, проводится поиск BLAST против всех белков выбранного организма (порог E-value = 10−5)[8] и пользователю предлагается выбрать одну из находок, для которой будут показаны возможные взаимодействия с другими белками (Proteins-режим) или COGs (COGs-режим) в данном организме.
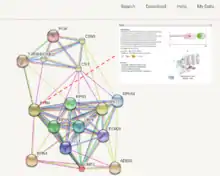
Экспериментально известные и предсказанные de novo взаимодействия заданного белка с другими белками представлены в виде графа, вершинами которого являются белки, а ребрами — различные типы доказательств функциональных взаимосвязей между этими белками. Вершины, соответствующие белкам, для которых расшифрована (или предсказана с определенной идентичностью) кристаллографическая структура, показаны более крупно. При клике на вершину во всплывающем окне доступны ссылки на сторонние ресурсы с информацией о данном белке, такие, как RefSeq, KEGG, UniProt, SMART и SWISS-MODEL, а также доступен предпросмотр доменной архитектуры и кристаллографической структуры (расшифрованной или предсказанной с определенной идентичностью) данного белка. Возможна кластеризация сети взаимодействий, добавление в сеть взаимодействий других белков при понижении порога достоверности функциональной взаимосвязи (и наоборот, удаление из сети взаимодействий белков при повышении порога), настройка допустимых типов доказательств функциональной взаимосвязи (например, можно оставить в сети взаимодействий только те белки, для взаимодействий которых есть экспериментальные доказательства), а также сохранение списка найденных белок-белковых взаимодействий в виде текстового файла и сохранение картинки сети взаимодействий[3].
Список возможных функциональных взаимосвязей заданного белка содержит доказательства каждой взаимосвязи и ранжирован по уровню оцененной достоверности каждой взаимосвязи[2].
Доступен просмотр филогенетического дерева, построенного по сцепленным выравниваниям последовательностей небольшого числа универсальных белковых семейств[5][15], с нанесёнными на него различными типами геномного контекста. Доступны ссылки на статьи, в которых упоминается заданный белок, в том числе экспериментальные статьи.
Интеграция с другими ресурсами
Имеется плагин STRING для Cytoscape[16]. Начиная с версии 10 (2015) программный пакет STRINGdb доступен для скачивания с Bioconductor и позволяет делать запросы к серверу STRING из языка программирования R[1].
Примечания
- D. Szklarczyk at al. STRING v10: protein–protein interaction networks, integrated over the tree of life (англ.) // Nucleic acids research : journal. — 2015. — Vol. 43. — P. D447—D452. — doi:10.1093/nar/gku1003. — PMID 25352553.
- A. Franceschini at al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration (англ.) // Nucleic acids research : journal. — 2013. — Vol. 41. — P. D808—D815. — doi:10.1093/nar/gks1094. — PMID 23203871.
- D. Szklarczyk at al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored (англ.) // Nucleic acids research : journal. — 2011. — Vol. 39. — P. D561—D568. — doi:10.1093/nar/gkq973. — PMID 21045058.
- L. Jensen at al. STRING 8—a global view on proteins and their functional interactions in 630 organisms (англ.) // Nucleic acids research : journal. — 2009. — Vol. 37. — P. D412—D416. — doi:10.1093/nar/gkn760. — PMID 18940858.
- C. Von Mering at al. STRING 7—recent developments in the integration and prediction of protein interactions (англ.) // Nucleic acids research : journal. — 2007. — Vol. 35. — P. D358—D362. — doi:10.1093/nar/gkl825. — PMID 17098935.
- C. Von Mering at al. STRING: known and predicted protein–protein associations, integrated and transferred across organisms (англ.) // Nucleic acids research : journal. — 2005. — Vol. 33. — P. D433—D437. — doi:10.1093/nar/gki005. — PMID 15608232.
- C. Von Mering at al. STRING: a database of predicted functional associations between proteins (англ.) // Nucleic acids research : journal. — 2003. — Vol. 31. — P. 258—261. — doi:10.1093/nar/gkg034. — PMID 12519996.
- B. Snel at al. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene (англ.) // Nucleic acids research : journal. — 2000. — Vol. 28. — P. 3442—3444. — doi:10.1093/nar/28.18.3442. — PMID 10982861.
- M. Huynen et al. Predicting Protein Function by Genomic Context: Quantitative Evaluation and Qualitative Inferences (англ.) // Genome research : journal. — 2000. — Vol. 10. — P. 1204—1210. — doi:10.1101/gr.10.8.1204. — PMID 10958638.
- M. Galperin et al. Expanded microbial genome coverage and improved protein family annotation in the COG database (англ.) // Nucleic acids research : journal. — 2015. — Vol. 43. — P. D261—D269. — doi:10.1093/nar/gku1223. — PMID 25428365.
- S. Powell et al. eggNOG v4.0: nested orthology inference across 3686 organisms (англ.) // Nucleic acids research : journal. — 2014. — Vol. 42. — P. D231—D239. — doi:10.1093/nar/gkt1253. — PMID 24297252.
- M. Price et al. Operon formation is driven by co-regulation and not by horizontal gene transfer (англ.) // Genome research : journal. — 2005. — Vol. 15. — P. 809—819. — doi:10.1101/gr.3368805. — PMID 15930492.
- A. Enright et al. Protein interaction maps for complete genomes based on gene fusion events (англ.) // Nature : journal. — 1999. — Vol. 402. — P. 86—90. — doi:10.1038/47056. — PMID 10573422.
- T. Rattei et al. SIMAP—a comprehensive database of pre-calculated protein sequence similarities, domains, annotations and clusters (англ.) // Nucleic acids research : journal. — 2010. — Vol. 38. — P. D223—D226. — doi:10.1093/nar/gkp949. — PMID 19906725.
- F. Ciccarelli et al. Toward Automatic Reconstruction of a Highly Resolved Tree of Life (англ.) // Science : journal. — 2006. — Vol. 311. — P. 1283—1287. — doi:10.1126/science.1123061. — PMID 16513982.
- Cytoscape. STRINGApp.