Орнитинтранскарбамилаза
Орнитинтранскарбамилаза (OTC) (также называемая орнитинкарбамоилтрансферазой) представляет собой фермент (Шифр КФ 2.1.3.3), который катализирует реакцию между карбамоилфосфатом (CP) и орнитином (Orn) с образованием цитруллина (Cit) и фосфата (Pi). Существует два класса OTC. Эта статья посвящена анаболическим OTC. Анаболические OTC обеспечивают шестой этап биосинтеза аминокислоты аргинина у прокариот[1]. Также OTC млекопитающих играют важную роль в цикле мочевины, целью которого является улавливание токсичного аммиака и превращение его в мочевину, менее токсичный источник азота, для выведения.
Механизм реакции
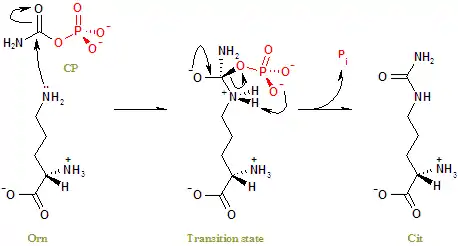
Структура
OTC — это тримерный белок. Есть три активных центра белка, которые расположены в пространстве между мономерами. Связывающий домен карбамоилфосфата находится на N-конце каждого мономера, в то время как C-конец содержит связывающий домен для орнитина. Оба связывающих домена имеют схожую структуру с центральным параллельным β-складчатым листом, ограниченным α-спиралями и петлями[3]. Помимо связывающих доменов, у OTC есть петли SMG. Они закрываются, закрывая сайт связывания, как только оба субстрата связываются. SMG означает консервативный аминокислотный мотив Ser-Met-Gly. После закрытия эти остатки взаимодействуют с L-орнитином. Связывание CP вызывает глобальные конформационные изменения, в то время как связывание L-орнитина только вызывает движение петли SMG для закрытия и изоляции сайта активации[4].
Активный сайт
Мотив Ser-Thr-Arg-Thr-Arg из одной субъединицы и His из соседней субъединицы взаимодействуют с фосфатной группой CP для связывания. Первичный азот CP связывается через остатки Gln, Cys и Arg. Карбонильный кислород CP связан остатками Thr, Arg и His[6].
Аминокислотный состав
OTC растений имеют наибольшее отличие от других ОТС. У них на 50-70 % меньше остатков Leu, в то время как остатков Arg в два раза больше. Количество аминокислот в ОТС варьируется от 322 до 340 остатков. Животные имеют самую высокую плотность Leu, которые обеспечивают pI для животного фермента 6,8, в то время как у растительного фермента pI составляет 7,6[7]. ОТС крыс, крупного рогатого скота и человека содержат один и тот же C-концевой остаток фенилаланина. С другой стороны, их N-концевые остатки различаются. У крыс в конце Ser, у быка — Asp, у человека — Gly[8][9].
Геномика
Ген ОТС человека расположен на коротком плече хромосомы X (Xp21.1). Ген расположен в (плюсовой) цепи Ватсона и имеет длину 73 т.п.н. Открытая рамка считывания из 1062 нуклеотидов распределяется между 10 экзонами и девятью интронами. Кодируемый белок имеет длину 354 аминокислоты с прогнозируемой молекулярной массой 39,935 кДа. Посттранскрипционная модификация оставляет зрелый пептид с 322 аминокислотами и массой 36,1 кДа[10]. Белок находится в митохондриальном матриксе. У млекопитающих ОТС экспрессируется в печени и слизистой оболочке тонкого кишечника.
Человеческие мутации
Известно о 341 мутации в человеческих безрецептурных препаратах. 149 из этих мутаций вызывают гипераммониемию в течение первых недель жизни. 70 проявляется гипераммониемией у пациентов мужского пола в более позднем возрасте. Большинство мутаций происходит в известных функциональных мотивах, таких как петля SMG или домены связывания CP[11].
Дефицит
Мутации в гене OTC могут вызывать дефицит орнитинтранскарбамилазы. Это заболевание классифицируется как нарушение цикла мочевины из-за того, что без обеспечения функции OTC аммиак начинает накапливаться в крови. Накопление аммиака в крови называется гипераммониемией. Поскольку аммиак, хотя и токсичен, но является источником азота для организма, его повышенный уровень вызывает повышение уровня аминокислот, глутамата и аланина. Уровни карбамоилфосфата (CP) начнут падать по мере снижения уровня азота мочевины в крови. Это приведет к тому, что CP переключится на путь синтеза уридинмонофосфата. Оротовая кислота является продуктом этого пути. Повышенный уровень оротовой кислоты в моче может быть индикатором того, что пациент страдает заболеванием, связанным с гипераммониемией.
Дефицит OTC проявляется как в ранней форме, так и в поздней форме.
Ранний дефицит
Ранний дефицит наблюдается у новорожденных. Симптомы нарушения цикла мочевины часто не проявляются до тех пор, пока ребёнок не окажется дома, и не могут быть своевременно обнаружены семьей и лечащим врачом . Симптомы у маленьких детей с гипераммониемией неспецифичны: нежелание есть, проблемы с дыханием, температурой тела, судороги, необычные движения тела (подергивания) и сонливость.[12] По мере накопления аммиака симптомы прогрессируют от сонливости до летаргии, потенциально заканчивающейся комой . Аномальная поза (неконтролируемое движение) и энцефалопатия (повреждение головного мозга) часто связаны со степенью отека центральной нервной системы и давления на ствол мозга. Приблизительно у 50 % новорожденных с тяжелой гипераммониемией возникают судороги.
Поздний дефицит
При более легкой (или частичной) недостаточности ферментов цикла мочевины накопление аммиака может быть вызвано болезнью или стрессом практически в любое время жизни, что приводит к многократному умеренному повышению концентрации аммиака в плазме [Bourrier et al. 1988]. У пациентов с частичным дефицитом ферментов симптомы могут проявляться с задержкой на месяцы или годы. Признаки того, что вы, возможно, страдаете от безрецептурного дефицита или нарушения цикла мочевины, включают «эпизоды бреда, беспорядочное поведение или подавленное сознание, головные боли, рвоту, отвращение к продуктам с высоким содержанием белка и судороги.»[13]
Лечение
Потенциальным средством лечения высокого уровня аммиака является введение бензоата натрия, который соединяется с глицином с образованием гиппурата с одновременным удалением группы аммония. Биотин также играет важную роль в функционировании OTC[14] и в экспериментах на животных было показано, что он снижает интоксикацию аммиаком. Кроме того, было предложено и изучено использование терапевтической гипотермии всего тела в качестве лечения. Считается, что она увеличивает эффективность диализа для извлечения аммиака из организма[15].
Примечания
- “Biosynthesis and metabolism of arginine in bacteria”. Microbiological Reviews. 50 (3): 314—52. September 1986. PMID 3534538.
- “Mechanism of inactivation of ornithine transcarbamoylase by Ndelta -(N'-Sulfodiaminophosphinyl)-L-ornithine, a true transition state analogue? Crystal structure and implications for catalytic mechanism”. The Journal of Biological Chemistry. 275 (26): 20012—9. June 2000. DOI:10.1074/jbc.M000585200. PMID 10747936.
- “The crystal structures of ornithine carbamoyltransferase from Mycobacterium tuberculosis and its ternary complex with carbamoyl phosphate and L-norvaline reveal the enzyme's catalytic mechanism”. Journal of Molecular Biology. 375 (4): 1052—63. January 2008. DOI:10.1016/j.jmb.2007.11.025. PMID 18062991.
- “Substrate-induced conformational change in a trimeric ornithine transcarbamoylase”. Proceedings of the National Academy of Sciences of the United States of America. 94 (18): 9550—5. September 1997. DOI:10.1073/pnas.94.18.9550. PMID 9275160.
- PDB 1C9Y; “Crystal structure of human ornithine transcarbamylase complexed with carbamoyl phosphate and L-norvaline at 1.9 A resolution”. Proteins. 39 (4): 271—7. June 2000. DOI:10.1002/(SICI)1097-0134(20000601)39:4<271::AID-PROT10>3.0.CO;2-E. PMID 10813810.
- “Human ornithine transcarbamylase: crystallographic insights into substrate recognition and conformational changes”. The Biochemical Journal. 354 (Pt 3): 501—9. March 2001. DOI:10.1042/bj3540501. PMID 11237854.
- “Purification and characterization of ornithine transcarbamylase from pea (Pisum sativum L.)”. Plant Physiology. 96 (1): 262—8. 1991-05-01. DOI:10.1104/pp.96.1.262. PMID 11538003.
- “Isolation and characterization of ornithine transcarbamylase from normal human liver”. The Journal of Biological Chemistry. 253 (11): 3939—44. June 1978. PMID 25896.
- “Ornithine transcarbamylase of rat liver. Kinetic, physical, and chemical properties”. The Journal of Biological Chemistry. 254 (20): 10030—6. October 1979. PMID 489581.
- “Targeting of pre-ornithine transcarbamylase to mitochondria: definition of critical regions and residues in the leader peptide”. Cell. 44 (3): 451—9. February 1986. DOI:10.1016/0092-8674(86)90466-6. PMID 3943133.
- “Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene”. Human Mutation. 27 (7): 626—32. July 2006. DOI:10.1002/humu.20339. PMID 16786505.
- Ornithine transcarbamylase deficiency. Genetics Home Reference. National Library of Medicine, U.S. Department of Health & Human Services. Дата обращения: 3 марта 2019.
- Ornithine transcarbamylase deficiency. Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. National Institutes of Health, U.S. Department of Health & Human Services. Дата обращения: 3 марта 2019.
- “Effect of biotin on ammonia intoxication in rats and mice”. Journal of Gastroenterology. 30 (3): 351—5. June 1995. DOI:10.1007/bf02347511. PMID 7647902.
- “Feasibility of adjunct therapeutic hypothermia treatment for hyperammonemia and encephalopathy due to urea cycle disorders and organic acidemias”. Molecular Genetics and Metabolism. 109 (4): 354—9. August 2013. DOI:10.1016/j.ymgme.2013.05.014. PMID 23791307.
Дальнейшее чтение
- Tuchman M, Plante RJ (1995). “Mutations and polymorphisms in the human ornithine transcarbamylase gene: mutation update addendum”. Human Mutation. 5 (4): 293—5. DOI:10.1002/humu.1380050404. PMID 7627182.
- Tuchman M (1993). “Mutations and polymorphisms in the human ornithine transcarbamylase gene”. Human Mutation. 2 (3): 174—8. DOI:10.1002/humu.1380020304. PMID 8364586.
- Matsuda I, Tanase S (September 1997). “The ornithine transcarbamylase (OTC) gene: mutations in 50 Japanese families with OTC deficiency”. American Journal of Medical Genetics. 71 (4): 378—83. DOI:10.1002/(SICI)1096-8628(19970905)71:4<378::AID-AJMG2>3.0.CO;2-Q. PMID 9286441.
- Wakabayashi Y (July 1998). “Tissue-selective expression of enzymes of arginine synthesis”. Current Opinion in Clinical Nutrition and Metabolic Care. 1 (4): 335—9. DOI:10.1097/00075197-199807000-00004. PMID 10565370.
- Tuchman M, Jaleel N, Morizono H, Sheehy L, Lynch MG (February 2002). “Mutations and polymorphisms in the human ornithine transcarbamylase gene”. Human Mutation. 19 (2): 93—107. DOI:10.1002/humu.10035. PMID 11793468.
- Feldmann D, Rozet JM, Pelet A, Hentzen D, Briand P, Hubert P, Largilliere C, Rabier D, Farriaux JP, Munnich A (July 1992). “Site specific screening for point mutations in ornithine transcarbamylase deficiency”. Journal of Medical Genetics. 29 (7): 471—5. PMC 1016021. PMID 1353535.
- Tuchman M, Holzknecht RA, Gueron AB, Berry SA, Tsai MY (November 1992). “Six new mutations in the ornithine transcarbamylase gene detected by single-strand conformational polymorphism”. Pediatric Research. 32 (5): 600—4. DOI:10.1203/00006450-199211000-00024. PMID 1480464.
- Dawson SJ, White LA (May 1992). “Treatment of Haemophilus aphrophilus endocarditis with ciprofloxacin”. The Journal of Infection. 24 (3): 317—20. DOI:10.1016/S0163-4453(05)80037-4. PMID 1602151.
- Suess PJ, Tsai MY, Holzknecht RA, Horowitz M, Tuchman M (June 1992). “Screening for gene deletions and known mutations in 13 patients with ornithine transcarbamylase deficiency”. Biochemical Medicine and Metabolic Biology. 47 (3): 250—9. DOI:10.1016/0885-4505(92)90033-U. PMID 1627356.
- Grompe M, Caskey CT, Fenwick RG (February 1991). “Improved molecular diagnostics for ornithine transcarbamylase deficiency”. American Journal of Human Genetics. 48 (2): 212—22. PMC 1683033. PMID 1671317.
- Hentzen D, Pelet A, Feldman D, Rabier D, Berthelot J, Munnich A (December 1991). “Fatal hyperammonemia resulting from a C-to-T mutation at a MspI site of the ornithine transcarbamylase gene”. Human Genetics. 88 (2): 153—6. DOI:10.1007/bf00206063. PMID 1721894.
- Strautnieks S, Rutland P, Malcolm S (December 1991). “Arginine 109 to glutamine mutation in a girl with ornithine carbamoyl transferase deficiency”. Journal of Medical Genetics. 28 (12): 871—4. DOI:10.1136/jmg.28.12.871. PMC 1017166. PMID 1757964.
- Carstens RP, Fenton WA, Rosenberg LR (June 1991). “Identification of RNA splicing errors resulting in human ornithine transcarbamylase deficiency”. American Journal of Human Genetics. 48 (6): 1105—14. PMC 1683104. PMID 2035531.
- Hata A, Matsuura T, Setoyama C, Shimada K, Yokoi T, Akaboshi I, Matsuda I (May 1991). “A novel missense mutation in exon 8 of the ornithine transcarbamylase gene in two unrelated male patients with mild ornithine transcarbamylase deficiency”. Human Genetics. 87 (1): 28—32. DOI:10.1007/BF01213087. PMID 2037279.
- Legius E, Baten E, Stul M, Marynen P, Cassiman JJ (August 1990). “Sporadic late onset ornithine transcarbamylase deficiency in a boy with somatic mosaicism for an intragenic deletion”. Clinical Genetics. 38 (2): 155—9. DOI:10.1111/j.1399-0004.1990.tb03565.x. PMID 2208768.
- Finkelstein JE, Francomano CA, Brusilow SW, Traystman MD (June 1990). “Use of denaturing gradient gel electrophoresis for detection of mutation and prospective diagnosis in late onset ornithine transcarbamylase deficiency”. Genomics. 7 (2): 167—72. DOI:10.1016/0888-7543(90)90537-5. PMID 2347583.
- Grompe M, Muzny DM, Caskey CT (August 1989). “Scanning detection of mutations in human ornithine transcarbamoylase by chemical mismatch cleavage”. Proceedings of the National Academy of Sciences of the United States of America. 86 (15): 5888—92. Bibcode:1989PNAS...86.5888G. DOI:10.1073/pnas.86.15.5888. PMC 297736. PMID 2474822.
- Lee JT, Nussbaum RL (December 1989). “An arginine to glutamine mutation in residue 109 of human ornithine transcarbamylase completely abolishes enzymatic activity in Cos1 cells”. The Journal of Clinical Investigation. 84 (6): 1762—6. DOI:10.1172/JCI114360. PMC 304053. PMID 2556444.
- Chu TW, Eftime R, Sztul E, Strauss AW (June 1989). “Synthetic transit peptides inhibit import and processing of mitochondrial precursor proteins”. The Journal of Biological Chemistry. 264 (16): 9552—8. PMID 2722850.
- Hata A, Setoyama C, Shimada K, Takeda E, Kuroda Y, Akaboshi I, Matsuda I (July 1989). “Ornithine transcarbamylase deficiency resulting from a C-to-T substitution in exon 5 of the ornithine transcarbamylase gene”. American Journal of Human Genetics. 45 (1): 123—7. PMC 1683378. PMID 2741942.
- Urea Cycle Disorders Overview. University of Washington, Seattle (29 April 2003).