Фактор фон Виллебранда
Фактор фон Виллебранда (англ. von Willebrand factor, VWF) — гликопротеин плазмы крови, играющий важную роль в гемостазе, а именно обеспечивающий прикрепление тромбоцитов к участку повреждённого сосуда. Кодируется геном VWF, расположенным на 12-й хромосоме. Нехватка или дефекты фактора фон Виллебранда приводят к развитию болезни Виллебранда и многих других заболеваний, в числе которых тромботическая пурпура, синдром Гейде и гемолитико-уремический синдром.
| Фактор фон Виллебранда | |
|---|---|
 | |
| Обозначения | |
| Символы | VWF |
| АТХ | B02BD10 |
| Entrez Gene | 7450 |
| HGNC | 12726 |
| OMIM | 193400 |
| RefSeq | NM_000552 |
| UniProt | P04275 |
| Другие данные | |
| Локус | 12-я хр. , 12p13.3 |
Ген
Фактор фон Виллебранда кодируется геном VWF, расположенным на коротком плече 12-й хромосомы в локусе 13.3 (с 5 948 874-й по 6 124 670-ю пару оснований). Известно более 300 мутаций гена VWF, приводящих к болезни Виллебранда[1].
Биохимия
Синтез
VWF — крупный мультимерный гликопротеин плазмы крови, который постоянно производится в виде ультра-крупных мультимеров клетками эндотелия (в тельцах Вайбеля — Паладе), мегакариоцитами (α-гранулы тромбоцитов) и субэндотелиальной соединительной тканью[2].
Структура
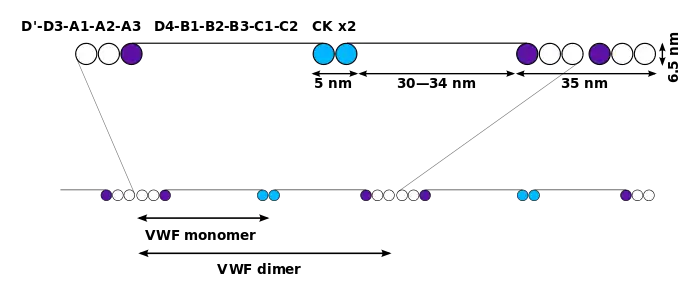
Мономер VWF представляет собой белок, состоящий из 2050 аминокислотных остатков. Каждый мономер содержит ряд доменов, выполняющих специфические функции; среди них стоит особо выделить:
- домен D'/D3 (домен типа D фактора фон Виллебранда), который связывается с фактором свёртывания крови VIII;
- домен А1, который связывается с гликопротеином Ib тромбоцитов, гепарином и, возможно, коллагеном;
- домен А3 (домен типа A фактора фон Виллебранда), который связывается с коллагеном;
- домен С1, в котором мотив аргинин-глицин-аспартат (RGD) связывается с тромбоцитарным интегрином αIIbβ3 при активации тромбоцитов;
- домен «цистеиновый узел», располагается на С-конце белка (домен типа C фактора фон Виллебранда). Такой домен имеется также у тромбоцитарного фактора роста (PDGF), трансформирующего фактора роста β (TGFβ) и человеческого хорионического гонадотропина β (βHCG)[2].
После синтеза мономеры подвергаются N-гликозилированию, собираются в димеры в эндоплазматическом ретикулуме и в мультимеры в аппарате Гольджи путём образования дисульфидных мостиков между остатками цистеина. Димеризацию осуществляют белковые дисульфидизомеразы[3]. VWF — один из немногих белков, несущих антигены системы групп крови ABO[2].
Мультимеры VWF могут быть очень крупными: состоять из более чем 80 субъединиц массой 250 кДа каждая и иметь массу более 20 000 кДа. Функциональны только крупные мультимеры[2].
Катаболизм
Биологическое разрушение (катаболизм) фактора фон Виллебранда осуществляет в основном фермент ADAMTS13 — металлопротеиназа, которая разрезает VWF между остатками тирозина 842 и метионина 843 в домене А2. Это приводит к распаду мультимеров на мелкие составляющие, которые разрушаются другими пептидазами[4].
Функции
Фактор фон Виллебранда играет важную роль в прикреплении тромбоцитов к местам повреждения сосудов, связываясь с другими белками, прежде всего фактором свёртывания крови VIII. Фактор свёртывания крови VIII связан с VWF, когда в неактивном состоянии циркулирует по кровотоку, и быстро разрушается, когда не связан с VWF. Связь фактора VIII с VWF разрушается под действием тромбина. Кроме того, VWF связывается с коллагеном (типа 1 альфа 1[5]), в том числе тогда, когда он соприкасается с эндотелиальными клетками в результате повреждения сосуда. Показано, что в связывании VWF с коллагеном имеет место эффект кооперативности[6]. VWF связывается с гликопротеином Ib, когда он формирует комплекс с гликопротеинами IX и V. Это связывание может происходить в любых условиях, однако оно наиболее сильно в условиях сильного напряжения сдвига, то есть при быстром движении крови в узких сосудах. Наконец, VWF связывается с другими рецепторами тромбоцитов, когда они активированы, например, тромбином (то есть когда уже произошла стимуляция коагуляции)[2].
Таким образом, VWF играет важную роль в свёртывании (коагуляции) крови, поэтому его недостаток или дисфункция увеличивает склонность к кровотечениям, особенно в тканях, в которых наблюдается высокая скорость кровотока в узких сосудах. В этих условиях VWF разворачивается, уменьшая скорость движения тромбоцитов[2]. Скорость рефолдинга домена А2 VWF увеличивается в присутствии ионов кальция, благодаря чему VWF может функционировать как сенсор напряжения сдвига[7].
Клиническое значение
Наследственные или приобретённые дефекты фактора фон Виллебранда приводят к болезни Виллебранда — геморрагическому диатезу кожи и слизистых оболочек, который выражается в носовых кровотечениях, меноррагии и желудочно-кишечных кровотечениях. Точка возникновения мутации определяет степень выраженности симптомов геморрагического диатеза[8]. Выделяют три типа болезни Виллебранда (I, II и III), а тип II, в свою очередь, делится на несколько подтипов. Большая часть случаев болезни Виллебранда носит наследственный характер, однако болезнь может быть и приобретённой. Так, стеноз аортального клапана связан с болезнью Виллебранда типа IIA и потому приводит к желудочно-кишечным кровотечениям; такое связанное заболевание получило название синдрома Гейде (англ. Heyde's syndrome)[9].
При тромботической пурпуре и уремическо-гемолитическом синдроме наблюдается недостаток фермента ADAMTS13 или его подавление антителами. Это приводит к снижению разрушения ультра-крупных мультимеров VWF и микроангиопатической гемолитической анемии, при которой в узких сосудах накапливаются фибрин и тромбоциты, из-за чего происходит некроз капилляров. При тромботической пурпуре поражается в основном мозг, а при уремико-гемолитическом синдроме — почки[10].
Высокий уровень VWF в крови характерен для людей, которые пережили первый ишемический инсульт в результате сворачивания крови. ADAMTS13 с этим не связана, и единственным значимым генетическим фактором в случае такого инсульта может быть группа крови пациента[11].
История изучения
Фактор назван по имени финского врача Эрика Адольфа фон Виллебранда (1870—1949), который в 1924 году описал наследственное заболевание крови (позже ставшее известным как болезнь Виллебранда) в нескольких семьях с Аландских островов. Члены этих семей имели склонность к кровотечениям из кожи и слизистых (включая меноррангию). Хотя фон Виллебранд не смог установить причину болезни, он сумел отличить её от гемофилии и других форм геморрагических диатезов[12]. В 1950-х годах было показано, что болезнь Виллебранда обусловлена нехваткой фактора плазмы крови, а не нарушением функционирования тромбоцитов, а в 1970-х годах был выделен фактор фон Виллебранда[2].
Примечания
- Genetics Home Reference: VWF.
- Sadler J. E. Biochemistry and genetics of von Willebrand factor. (англ.) // Annual review of biochemistry. — 1998. — Vol. 67. — P. 395—424. — doi:10.1146/annurev.biochem.67.1.395. — PMID 9759493.
- Lippok S., Kolsek K., Löf A., Eggert D., Vanderlinden W., Müller J. P., König G., Obser T., Röhrs K., Schneppenheim S., Budde U., Baldauf C., Aponte-Santamaría C., Gräter F., Schneppenheim R., Rädler J. O., Brehm M. A. Von Willebrand factor is dimerized by protein disulfide isomerase. (англ.) // Blood. — 2015. — doi:10.1182/blood-2015-04-641902. — PMID 26670633.
- Levy G. G., Motto D. G., Ginsburg D. ADAMTS13 turns 3. (англ.) // Blood. — 2005. — Vol. 106, no. 1. — P. 11—17. — doi:10.1182/blood-2004-10-4097. — PMID 15774620.
- Pareti F. I., Fujimura Y., Dent J. A., Holland L. Z., Zimmerman T. S., Ruggeri Z. M. Isolation and characterization of a collagen binding domain in human von Willebrand factor. (англ.) // The Journal of biological chemistry. — 1986. — Vol. 261, no. 32. — P. 15310—15315. — PMID 3490481.
- Heidari M., Mehrbod M., Ejtehadi M. R., Mofrad M. R. Cooperation within von Willebrand factors enhances adsorption mechanism. (англ.) // Journal of the Royal Society, Interface / the Royal Society. — 2015. — Vol. 12, no. 109. — P. 20150334. — doi:10.1098/rsif.2015.0334. — PMID 26179989.
- Jakobi A. J., Mashaghi A., Tans S. J., Huizinga E. G. Calcium modulates force sensing by the von Willebrand factor A2 domain. (англ.) // Nature communications. — 2011. — Vol. 2. — P. 385. — doi:10.1038/ncomms1385. — PMID 21750539.
- Sadler J. E., Budde U., Eikenboom J. C., Favaloro E. J., Hill F. G., Holmberg L., Ingerslev J., Lee C. A., Lillicrap D., Mannucci P. M., Mazurier C., Meyer D., Nichols W. L., Nishino M., Peake I. R., Rodeghiero F., Schneppenheim R., Ruggeri Z. M., Srivastava A., Montgomery R. R., Federici A. B. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. (англ.) // Journal of thrombosis and haemostasis : JTH. — 2006. — Vol. 4, no. 10. — P. 2103—2114. — doi:10.1111/j.1538-7836.2006.02146.x. — PMID 16889557.
- Vincentelli A., Susen S., Le Tourneau T., Six I., Fabre O., Juthier F., Bauters A., Decoene C., Goudemand J., Prat A., Jude B. Acquired von Willebrand syndrome in aortic stenosis. (англ.) // The New England journal of medicine. — 2003. — Vol. 349, no. 4. — P. 343—349. — doi:10.1056/NEJMoa022831. — PMID 12878741.
- Moake J. L. von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. (англ.) // Seminars in hematology. — 2004. — Vol. 41, no. 1. — P. 4—14. — PMID 14727254.
- Bongers T. N., de Maat M. P., van Goor M. L., Bhagwanbali V., van Vliet H. H., Gómez Garc E. B., Dippel D. W., Leebeek F. W. High von Willebrand factor levels increase the risk of first ischemic stroke: influence of ADAMTS13, inflammation, and genetic variability. (англ.) // Stroke; a journal of cerebral circulation. — 2006. — Vol. 37, no. 11. — P. 2672—2677. — doi:10.1161/01.STR.0000244767.39962.f7. — PMID 16990571.
- Von Willebrand E. A. Hereditary pseudohaemophilia. (англ.) // Haemophilia : the official journal of the World Federation of Hemophilia. — 1999. — Vol. 5, no. 3. — P. 223—231. — PMID 10444294.
Литература
- Blackshear J. L., Kusumoto H., Safford R. E., Wysokinska E., Thomas C. S., Waldo O. A., Stark M. E., Shapiro B. P., Ung S., Moussa I., Agnew R. C., Landolfo K., Chen D. Usefulness of Von Willebrand Factor Activity Indexes to Predict Therapeutic Response in Hypertrophic Cardiomyopathy. (англ.) // The American journal of cardiology. — 2015. — doi:10.1016/j.amjcard.2015.11.016. — PMID 26705879.
- Hudzik B., Kaczmarski J., Pacholewicz J., Zakliczynski M., Gasior M., Zembala M. Von Willebrand factor in patients on mechanical circulatory support - a double-edged sword between bleeding and thrombosis. (англ.) // Kardiochirurgia i torakochirurgia polska = Polish journal of cardio-thoracic surgery. — 2015. — Vol. 12, no. 3. — P. 233—237. — doi:10.5114/kitp.2015.54459. — PMID 26702279.
- Li Y., Li L., Dong F., Guo L., Hou Y., Hu H., Yan S., Zhou X., Liao L., Allen T. D., Liu J. U. Plasma von Willebrand factor level is transiently elevated in a rat model of acute myocardial infarction. (англ.) // Experimental and therapeutic medicine. — 2015. — Vol. 10, no. 5. — P. 1743—1749. — doi:10.3892/etm.2015.2721. — PMID 26640545.