Миелодиспластический синдром
Миелодиспластический синдром (МДС) — группа гетерогенных клональных заболеваний, характеризующаяся наличием цитопении в периферической крови, дисплазии в костном мозге и риском трансформации в острый лейкоз.
| Миелодиспластический синдром | |
|---|---|
| МКБ-11 | XH7PK9 |
| МКБ-10 | D46 |
| МКБ-9 | 238.7 |
| МКБ-9-КМ | 238.75[1] и 238.7[1] |
| МКБ-О | 9980/0-M9989/3 |
| OMIM | 614286 |
| DiseasesDB | 8604 |
| MedlinePlus | 007716 |
| eMedicine | med/2695 ped/1527 |
| MeSH | D009190 |
МДС сегодня является одной из самых сложных проблем гематологии. Лишь недавно лечение МДС вышло за рамки поддерживающей терапии, проводившейся с целью облегчения симптомов.
МДС является патологией старшей возрастной группы: 80 % случаев МДС приходится на лица старше 60 лет. МДС в детском возрасте встречается крайне редко. В европейских странах среди лиц 50—69 лет регистрируется 40 новых случаев МДС на 1 млн населения, а среди лиц 70 лет и старше — 150 новых случаев на 1 млн населения. Заболеваемость МДС в РФ в среднем составляет 3—4 случая на 100 тыс. населения в год и увеличивается с возрастом.[2]
Типы, факторы риска
Первичный (идиопатический) тип — 80—90 % случаев, вторичный (вследствие предшествующей химиотерапии и др. факторов) — 10—20 %. Большинство (80 %) случаев МДС являются первичными — идиопатическими или de novo (с лат. — «вновь появившийся, новый»).
Вторичный МДС является значительно более неблагоприятным и резистентным к лечению типом МДС, обладающим заведомо более худшим прогнозом в сравнении с первичным МДС. 10—20 % случаев МДС возникают вследствие предшествующей химиотерапии по поводу других новообразований. К препаратам, обладающим доказанной способностью повреждать геном с последующим развитием МДС, относятся алкилирующие агенты (циклофосфан), ингибиторы топоизомеразы — противоопухолевые агенты растительного происхождения (топотекан, иринотекан и др.), антрациклины (доксорубицин) и подофиллотоксины (этопозид). К МДС также могут приводить радиотерапия и контакт с токсическими материалами.
- Факторы риска, первичный МДС
- Контакт с токсинами (бензин, органические растворители, в частности бензол, пестициды)
- Радиация
- Курение
- Врождённые и наследственные заболевания
- Пожилой возраст
- Факторы риска, вторичный МДС
Предшествующая химиотерапия онкологического заболевания или после ТКМ.
Прогноз: 5-летняя выживаемость при МДС не превышает 60 %. Трансформация в острый лейкоз в около 30 % случаев.[3][4]
Патогенез
Причины МДС до конца не известны. В основе патогенеза МДС лежит воздействие повреждающих факторов на полипотентную стволовую клетку, приводящее к появлению в ней генетических аномалий, а также феномена гиперметилирования ДНК.
Указанные нарушения приводят к нарушению продукции клеток миелоидного ростка и появлению миелобластов в костном мозге и периферической крови, вследствие чего появляются диспластические изменения в зрелых клетках и их функциональная недостаточность, приводящие к описанным клиническим проявлениям.
Феномен гиперклеточности костного мозга на фоне периферической цитопении объясняется ускоренным апоптозом аномально пролиферирующих клеток костного мозга.[5]
Клиническая картина
МДС отличает отсутствие типичной клинической картины. Симптоматику МДС составляют последствия дисмиелопоэза, то есть цитопении: анемия, нейтропения и тромбоцитопения (анемия Hb меньше 110 г/л, нейтрофилы меньше 1800 на 1 микролитр крови; гематокрит меньше 36 % эритроцитов в общем объёме крови в организме; тромбоциты меньше 100 000 на 1 микролитр крови).
Наиболее часто МДС манифестирует цитопениями, главным образом анемией. При этом необходимо дифференцировать МДС от железо- или B12-дефицитной анемии, постгеморрагической анемии, анемии при хронических заболеваниях и онкологии или связанной с хронической почечной недостаточностью, а также апластической анемией, пароксизмальной ночной гемоглобинурией. У 10 % пациентов имеются признаки инфекции, а у несколько меньшей доли пациентов болезнь проявляется кровотечениями.
В связи с этим диагностика МДС базируется исключительно на лабораторно-инструментальных методах, из которых ключевыми являются полный клинический анализ периферической крови, некоторые биохимические исследования и морфологический анализ аспиратов и биоптатов костного мозга.
Дифференциальная диагностика МДС также затруднена в силу множества состояний, имеющих общие с МДС клинико-лабораторные проявления.
План обследования пациентов
Для анализа крови изменений в периферической крови проводится полный, с подсчётом ретикулоцитов (ускоренный эритропоэз с макроцитозом в ответ на гемолиз и острую кровопотерю приводит к увеличению ретикулоцитов), тромбоцитов и лейкоцитов клинический анализ крови. Типичными находками являются изменения формы клеток, патологические включения и уменьшение числа клеток одного или нескольких ростков кроветворения.
Другим ключевым с точки зрения диагностики МДС оценки прогноза и выработки тактики лечения больных исследованием является морфологическое, иммуногистохимическое и цитогенетическое исследования ткани костного мозга. Исследование костномозгового пунктата в этом отношении является несравненно более информативным, чем определение морфологического состава периферической крови.
Используются два способа получения материала: аспирационная биопсия костного мозга и трепанобиопсия из гребня подвздошной кости.
При цитологическом исследовании костного мозга (миелограмма) можно оценить наличие дисплазии миелоидного ростка.
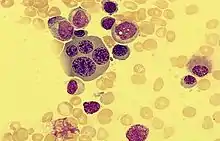

- Дизэритропоэз
- Межъядерные цитоплазматические мостики
- Кариорексис
- Многоядерность
- Баббл-формы
- Мегалобластоидность
- Кольцевые сидеробласты
- Вакуолизация
- Положительная реакция с Шифф-йодной кислотой
- Дисгранулопоэз
- Маленькие или чрезмерно большие клетки
- Гиполобулярность ядер (псевдо-Пельгер-Хюит аномалия)
- Неравномерная гиперсегментация
- Гипо-/агрануляция
- Гранулы псевдо Чедиак-Хагаси
- Палочки Ауэра
- Дисмегакариопоэз
- Микромегакариоциты
- Гиполобулярные ядра
- Многоядерность
Гистологическое исследование костного мозга (трепанобиопсия) позволяет оценить архитектонику костного мозга, диффузный или очаговый характер изменений в нём, изучить соотношение кроветворной и жировой ткани, выявить атипичные клетки и т. п. Аспирация костного мозга при стернальной пункции так или иначе нарушает структуру костного мозга и не исключает примешивание к пунктату периферической крови. В связи с этим выполнение трепанобиопсии обязательно для подтверждения диагноза МДС.
Биохимические исследования обмена железа, содержания витамина В12 и фолиевой кислоты, иммунологические пробы призваны помочь провести дифференциальную диагностику с анемиями иного генеза, с учётом того, что у 80 % пациентов с МДС отмечается анемия.
МДС следует дифференцировать с другими онкогематологическими заболеваниями, включая острые и хронические лейкозы, а также лимфопролиферативные заболевания.
Часть изменений, свойственных МДС (в частности, моноцитоз, цитопенические нарушения), могут отмечаться при некоторых инфекционных процессах.
При отравлении тяжелыми металлами могут отмечаться изменения эритроцитарного ростка, сходные с таковыми при сидеробластных анемиях.
У пациентов с наследственными цитопениями рекомендуется проведение дополнительного генетического исследования, которое поможет выявить анемию Фанкони и врождённый дискератоз.
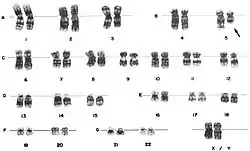
Цитогенетические нарушения

При диагностике хромосомные аномалии обнаруживаются у 40—70 % пациентов с первичным MDS и у 95 % пациентов, MDS которых связан с терапией (вторичный).
К наиболее часто встречающимся при MDS цитогенетическим аномалиям относятся del(5q), −7 и +8.[6]
| Группа риска | Кариотип (22 группы) | Средняя выживаемость (мес) | Время, к которому 25 % пациентов развили ОМЛ |
|---|---|---|---|
| Благоприятная | 5q−, 12p−, 20q−, +21, −Y, 11q−, t(11(q23)), норма; любые 2 аномалии, включающие 5q− | 51 | 71,9 |
| Промежуточная-1 | +1q, аномалии 3q21/q26, +8, t(7q), +19, −21, любая др. одиночная поломка; любые двойные аномалии, не затрагивающие хр. 5q и 7 | 29 | 16 |
| Промежуточная-2 | −X, −7 или 7q−, любые двойные аномалии с −7 или 7q−, комплекс из 3 аномалий | 15,6 | 6 |
| Неблагоприятная | Более 3 аномалий | 5,9 | 2,8 |
Минимальные диагностические критерии
Минимальные диагностические критерии МДС включают обязательные диагностические условия[7] — стабильная цитопения не менее 6 месяцев, (за исключением случаев, когда цитопения сопровождается специфическим кариотипом или дисплазией двух ростков кроветворения — в этих случаях длительность стабильной цитопении должна составлять не менее 2 месяцев).
- исключение других заболеваний, которые могут стать причиной развития дисплазии или/и цитопении.
В дополнение к этим двум диагностическим условиям для установления диагноза МДС необходимо соответствие хотя бы одному из трёх основных критериев:
- дисплазия (⩾ 10 % клеток одного или более из трёх основных ростков кроветворения в костном мозге).
- содержание бластов в костном мозге 5—19 %.
- специфический кариотип, например делеция (5q), делеция (20q), +8 или −7/делеция (7q).
Кроме того, для диагностики МДС используются дополнительные критерии, в том числе результаты проточной цитометрии, гистологического и иммуногистохимического исследования костного мозга, выявления молекулярных маркеров.
Морфологическое исследование биоптатов, полученных путём билатеральной трепанобиопсии, является полезным, помимо верификации диагноза самого МДС, с точки зрения дифференциальной диагностики с лимфопролиферативными и другими миелопролиферативными заболеваниями.[7]
Дифференциальная диагностика
Дифференциальная диагностика проводится с:
- Мегалобластными анемиями (заболевания, характеризующиеся изменениями морфологии клеток костного мозга вследствие нарушения синтеза ДНК. Более 90 % — В-12 и фолиево-дефицитные анемии). После начала терапии витамином В-12 или фолиевой кислотой в анализе крови выявляется ретикулярный криз на 5—7-е сутки и повышение показателей красной крови, что нехарактерно для больных рефрактерной анемией. Изменения кариотипа клеток костного мозга не встречаются при мегалобластных анемиях.
- Апластической анемией. Апластическая анемия может быть врождённой, приобретённой и идиопатической. Врождённая апластическая анемия — анемия Фанкони сочетается с другими генетическими аномалиями (кожная пигментация, гипоплазия почек, микроцефалия), приобретённая связана с действием химических и физических агентов, инфекциями, нарушениями обмена веществ. Для АА нехарактерны изменение кариотипа, гиперклеточный костный мозг.
- Анемии при ХПН.
- Анемии при хроническом активном гепатите характерно выявление маркеров вирусных инфекций, гепатоспленомегалия, клиническая картина хронического гепатита, изменения биохимических показателей крови (метаболизма билирубина, функции печени).
ФАБ-классификация
Разработка этой системы классификации франко-американо-британской группой была начата в 1976 году и позже, в 1982 году, она приняла свой окончательный вид.
В основе классификации лежит ключевой для МДС синдром — рефрактерная, то есть устойчивая к лечению препаратами витамина В12 и фолиевой кислоты, анемия (РА). Четыре типа РА являются последовательными стадиями, с нарастанием тяжести МДС, что имеет своё отражение в прогнозе выживаемости. В этой связи появление в КМ бластов резко меняет прогноз выживаемости в худшую сторону.
| Тип МДС | Бластов в периферической крови | Бластов в КМ | Другие патологические изменения | Выживаемость (лет) |
|---|---|---|---|---|
| Рефрактерная анемия (РА) | меньше 1 % | меньше 15 % кольцевых сидеробластов | меньше 5 % | 4,2 |
| РА с кольцевыми сидеробластами | меньше 1 % | больше 15 % кольцевых сидеробластов | меньше 5 % | 6,9 |
| РА с избытком бластов (РАИБ) | меньше 5 % | 5—20 % | — | 1,5 |
| РАИБ в стадии трансформации | больше 5 % | 21—29 % | Возможно наличие палочек Ауэра в КМ | 0,6 |
| ХММЛ | меньше 5 % | меньше 20 % | Моноциты больше 109/л | 2,4 |
Французско-американско-британская классификация позволяет отнести пациента к той или иной группе миелодиспластических синдромов в зависимости от морфологических показателей. Группа миелодиспластических синдромов включает пять заболеваний: рефрактерную анемию, рефрактерную анемию с кольцевыми сидеробластами, рефрактерную анемию с избыточным количеством бластов, рефрактерную анемию с избыточным количеством бластов на стадии трансформации и хронический миеломоноцитарный лейкоз. Согласно французско-американско-британской номенклатуре пациентам, у которых содержание бластов в костном мозге превышает 30 %, устанавливается диагноз острого миелоидного лейкоза.
В данной классификации хронический миеломоноцитарный лейкоз относится к группе миелодиспластических синдромов, несмотря на то, что это заболевание часто характеризуется признаками миелопролиферативного расстройства.[8]
Классификация ВОЗ
В 2002 году Всемирная организация здравоохранения предложила новую классификацию миелодиспластических синдромов[9][10][11] в 2008 году были сделаны предложения по её пересмотру.[12][13]
Подгруппы, выделяемые в классификации ВОЗ включают: рефрактерную анемию и рефрактерную анемию с кольцевыми сидеробластами, рефрактерную цитопению с множественной дисплазией, рефрактерную анемию с избыточным количеством бластов-1 (содержание бластов в костном мозге составляет менее 10 %), рефрактерную анемию с избыточным количеством бластов-2 (содержание бластов в костном мозге превышает 10 %), синдром делеции 5q и миелодиспластический синдром неклассифицированный (с наличием или отсутствием кольцевых сидеробластов).
Пациенты, ранее классифицировавшиеся как страдающие хроническим миеломоноцитарным лейкозом, относятся к группе миелодиспластических синдромов/миелопролиферативных заболеваний.
Синдром делеции 5q, выделяемый в классификации Всемирной организации здравоохранения в отдельную подгруппу, характеризуется изолированной делецией 5q[14][15][16] и содержанием бластов в костном мозге меньше 5 %, часто в сочетании с тромбоцитозом.
| Тип МДС | Изменения в крови | Изменения в КМ |
|---|---|---|
| Рефрактерная анемия (РА) | Анемия, меньше 1 % бластов | Дисплазия эритроидного ростка, меньше 5 % бластов |
| Рефрактерная анемия с кольцевыми сидеробластами (РАКС) | То же, что и РА | то же, что и РА, ⩾ 15 % кольцевых сидеробластов |
| Рефрактерная цитопения с многоростковой дисплазией (РЦМД) | Цитопения по 2—3 росткам, меньше 1 % бластов | Дисплазия в больше 10 % клеток 2 или 3 ростков, меньше 5 % бластов, меньше 15 % кольцевых сидеробластов |
| Рефрактерная цитопения с мультилинейной дисплазией и кольцевыми сидеробластами (РЦМД-КС) | То же, что и РЦМД | То же, что и РЦМД, ⩾ 15 % кольцевых сидеробластов |
| Рефрактерная анемия с избытком бластов, тип I (РАИБ-1) | Цитопении, меньше 5 % бластов | 5—9 % бластов |
| Рефрактерная анемия с избытком бластов, тип II (РАИБ-2) | Цитопении, 5—19 % бластов | 10—19 % бластов |
| Синдром 5q− | Анемия, нормальное или повышенное содержание тромбоцитов | Нормальное или увеличенное количество мегакариоцитов с гипосегментированными ядрами; изолированная делеция 5q31 |
| МДС неклассифицированный (МДС-Н) | Цитопения | Унилинейная дисплазия в нейтрофильном или мегакариоцитарном ростках, Бласты менее 5 %, Палочки Ауэра отсутствуют |
Всемирная организация здравоохранения предложила исключить рефрактерную анемию с избыточным количеством бластов на стадии трансформации из группы миелодиспластических синдромов (диагноз острого миелоидного лейкоза устанавливается, если содержание бластов в костном мозге превышает 20 %, тогда как ранее для установления этого диагноза содержание бластов должно было превышать 30 %). Однако миелодиспластические синдромы отличаются от вновь диагностированного острого миелоидного лейкоза не только содержанием бластов, но и течением заболевания, обусловленным определёнными биологическими свойствами. Кроме того, эти группы заболеваний обычно отличаются и по частоте терапевтических ответов.
Шкала IPSS

Шкала IPSS (International Scoring Prognostic System — Международная шкала оценки прогноза) была разработана в 1997 году с целью дать специалистам, помимо классификации, практический инструмент по оценке прогноза и выбора тактики лечения для пациентов с впервые установленным диагнозом МДС (то есть не подходит для прогноза уже леченных пациентов с МДС).
Вторичный МДС оценивается как изначально неблагоприятный, автоматически попадающий в категорию наиболее высокого риска согласно IPSS.
Тремя факторами, которые учитывает IPSS для оценки прогноза, являются количество бластов, категория цитогенетического риска и количество поражённых цитопенией линий.
Трактовка результатов суммирования баллов по этим трем параметрам:[16]
| Количество балов | ||||||
|---|---|---|---|---|---|---|
| Прогностический фактор | 0 | 0,5 | 1,0 | 1,5 | 2,0 | |
| Бласты в костном мозге | меньше 5 % | 5—10 % | — | 11—20 % | 21—30 % | |
| Прогноз с учётом характеристик кариотипа | Хороший (норма, del(5q) del(20q) −Y) | Промежуточный (+8 хромосома, 2 аномалии и др.) | Плохой (аномалии 7-й хромосомы, ⩾ 3 аномалии) | — | — | |
| Цитопения (количество поражённых линий) | 0/1 | 2/3 | — | — | — | |
Прогноз у пациентов
Сумма баллов, соответствующая высокому риску по IPSS (больше 2,5) складывается из мультилинейной дисплазии, плохого цитогенетического прогноза и высокого бластоза, на грани перехода в ОМЛ (срок трансформации в который в категории высокого риска составляет всего 2 месяца).
Категория промежуточного-2 риска также складывается из выраженного цитопенического синдрома и высокого, в пределах 10-20 % бластоза.
То, что в категории низкого риска медиана общей выживаемости ниже срока перехода в ОМЛ, объясняется меньшим сроком жизни больных с МДС, что отражает последствия осложнений цитопенического синдрома.[17]
| Сумма баллов | Риск по IPSS | Срок до перехода в ОМЛ у 25 % пациентов (лет) | Медиана общей выживаемости (лет) | Доля пациентов |
|---|---|---|---|---|
| 0 | Низкий | 9,4 | 5,7 | 31 % |
| 0,5—1,0 | Промежуточный-1 | 3,3 | 3,5 | 39 % |
| 1,5—2,0 | Промежуточный-2 | 1,1 | 1,2 | 22 % |
| ⩾ 2,5 | Высокий | 0,2 | 0,4 | 8 % |
Прогностическая система ВОЗ (WPSS)
| Баллы | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| Вид МДС по классификации ВОЗ | РА, РАКС, 5q− | РЦМД, РЦМД-КС | РАИБ1 | РАИБ2 |
| Кариотип | Хороший | Средний | Плохой | — |
| Потребность в гемотрансфузиях | Нет | Регулярная | — | — |
Кариотип:
- Хороший: нормальный, −Y, del 5q, del 20q.
- Плохой: более 3 аномалий или аномалии 7-й хромосомы.
- Средний: все другие.
Регулярные гемотрансфузии — переливание минимум 1 ЭМ каждые 8 недель в течение 4 месяцев.
| Группа риска | Баллы | Медиана выживаемости (мес) |
|---|---|---|
| Очень низкий | 0 | 136 |
| Низкий | 1 | 63 |
| Средний | 2 | 44 |
| Высокий | 3—4 | 19 |
| Очень высокий | 5—6 | 8 |
Методы лечения
Не все пациенты с МДС нуждаются в терапии. Пациенты без анемического, геморрагического синдрома, инфекционных осложнений могут наблюдаться и не получать лечения (тактика «watch and wait»).
Выбор терапевтической тактики во многом определяется возрастом пациента, соматическим статусом, степенью риска по шкале IPSS, WPSS, наличием совместимого донора.
Можно выделить следующие направления терапии МДС:
- Сопроводительная терапия включает в себя переливание различных гемокомпонентов (эритроцитарной массы, тромбоконцентрата), терапию эритропоэтином, тромбопоэтином. У больных, часто получающих гемотрансфузии, развивается перегрузка организма железом. Железо обладает токсическим действием на различные ткани и органы, в первую очередь сердце, печень, поэтому такие пациенты должны получать препараты, связывающие железо, — хелаторы (десферал, эксиджад).
- Иммуносупрессивная терапия наиболее эффективна у пациентов с гипоклеточным костным мозгом, нормальным кариотипом и наличием HLA-DR15. Леналидомид, обладающий иммуномодулирующим и антиангиогенным действием, показал свою эффективность у трети пациентов с рефрактерной анемией (согласно критериям ВОЗ) и низким риском (по IPSS), а также у больных с 5q-синдромом. Эффективность лечения в данном случае весьма высока; 95 % больных достигают цитогенетической ремиссии.
- Аллогенная трансплантация гемопоэтических стволовых клеток от совместимых доноров является методом выбора у пациентов с миелодиспластическим синдромом.[19][20][21] Пациентам с МДС моложе 65 лет, с хорошим соматическим статусом, при наличии HLA-совместимого донора показано проведение аллогенной трансплантации костного мозга, так как трансплантация является потенциально радикальным методом лечения МДС.
- Химиотерапия. Например, низкие дозы цитарабина широко используются в России и в Европе для лечения пациентов с МДС и ОМЛ, которым не подходит терапия методом ТКМ или применение интенсивной химиотерапии. Мнения исследователей относительно целесообразности использования низкоинтенсивной терапии расходятся. Дэвид Боуэн[22] считает, что нет оснований рекомендовать её рутинное использование при МДС: было выполнено 3 рандомизированных крупных исследования (141 пацицентов), которые показали, что применение низких доз цитарабина не увеличивает продолжительность жизни пациентов с МДС.[23] Вместе с тем, в более позднем исследовании у пациентов с ОМЛ и МДС высокого риска[24] было показано, что продолжительность жизни у больных, у которых применялся LDAC более, чем в 1 цикле, выше, чем при поддерживающей терапии. Таким образом, необходимость в низкоинтенсивной терапии с доказанной эффективностью и лучшей переносимостью, чем LDAC, которая будет способствовать увеличению выживаемости пациентов с МДС высокого риска, остаётся актуальной. Высокодозная химиотерапия используется у больных с РАИБ с гипер- и нормоклеточным костным, при трансформации в ОМЛ. Пятилетняя выживаемость составляет около 18 %.
- Гипометилирующие препараты. Новые многообещающие терапевтические подходы, широко обсуждающиеся в последнее время, по поводу которых проводятся многочисленные клинические исследования, возникли в результате глубокого изучения биологии МДС. Среди них следует отметить ингибиторы метилирования ДНК (5-азацитидин, децитабин) и иммуномодулятор — леналидомид. 5-азацитидин обладает двойным механизмом действия. Он встраивается не только в молекулу ДНК, но и в молекулу РНК. В процессе метилирования ДНК гипометилирующие агенты ковалентно связываются с ДНК-метилтрансферазой, что приводит к реактивации генов, после чего восстанавливается дифференцировка гемопоэтических клеток-предшественников и нормальное кроветворение. Азацитидин, встраиваясь в РНК молекулу, тем самым понижает её количество в клетках, что приводит к цитостатическому эффекту вне зависимости от клеточной фазы. На основании результатов исследования 3 фазы AZA-001 — международное, мультицентровое, контролируемое, в параллельных группах, в котором пациенты МДС высокого риска/ОМЛ (ВОЗ критерии) сравнивались со стандартным лечением (сопроводительная терапия, интенсивная химиотерапия, низкие дозы цитарабина), азацитидин был зарегистрирован, в том числе и в РФ, для лечения этих больных. Было показано, что азацитидин в 2,5 раза увеличивает общую выживаемость.
Примечания
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- Гематология / Под редак. О. А. Рукавицына. — СПб., 2007. — С. 193—226.
- Corey S. J., Minden M. D., Barber D. L. et al. Myelodysplastic syndromes: the complexicity of stem-cell diseases. Cancer. Nature reviews. 2007. V. 7; 118—129.
- Pedersen-Bjergaard J., Pedersen M., Roulston D., Philip P.. (1995) Different genetic pathways in leukemogenesis for patients presenting with therapy-related myelodysplasia and therapy-related acute myeloid leukemia. Blood. 86(9): 3542—3552.
- Greenberg P. L. Apoptosis and its role in the in the myelodysplastic syndromes; implications for disease natural history and treatment. Leuk res, 1998; 22: 1123—1136
- Onley H. J., Le Beau M. M. Cytogenetic Diagnosis of Myelodysplastic syndromes. in book H. J. Deeg, D. T. Bowen, S. D. Gore, T. Haferlach, m. M. Le Beau, C. Niemeyr. Hematologic Malignancies: Myelodysplastic syndromes // Springler Berlin Heidelbery. 2006, P. 55—79.
- NCCN Clinical Practice Guidelines in oncology. Myelodisplastic syndromes (англ.) : journal. — 2009. — Vol. 1. Архивировано 31 октября 2010 года.[прояснить]
- Bennett J. M., Catovsky D., Daniel M. T., Flandrin G., Galton D. A., et al. (1982) Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol. 51(2): 189—199.
- Brunning R., Bennett J., Flandrin G. et al. Myelodysplastic Syndromes. In: Jaffe E, Harris N, Stein H et al., eds. WHO Classification of Tumours. Pathology and Genetics of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press 2001; 61—73.
- Harris N., Jaffe E., Diebold J. et al. WHO Classification of Neoplastic Diseases of the Hematopoietic and Lymphoid Tissues: Report of the Clinical Advisory Committee Meeting. J. Clin. Oncol. 1999; 17:3835—3849.
- Vardiman J. W., Harris N. L., Brunning R. D. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002; 100:2292—2302.
- Bruning R. D., Orazi A., Germing U. et al., 2008. WHO classification of tumors of hematopoietic and lymphoid tissues. Chapter 5, p. 88—107.
- Hollstrem Lindberg E., Cazzola M. The role of JAK2 mutations in RARS and other MDS. 2008. Hematology, 52—59.
- Greenberg P. L., Baer M., Bennett J. et al. NCCN Practice Guidelines for Myelodysplastic Syndromes, Version1, 2001, In «The Complete Library of NCCN Guidelines [CD-ROM]», Rockledge, PA.
- Cheson B. D., Bennett J. M., Kantarjian H. et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood 2000; 96:3671—3674.
- Greenberg P., Cox c., Le Beau M. M. et al., International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89:2079—2088.
- Malcovati L., Germing U., Kuendgen A. et al. Time-dependant prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 2007; 25: 3503—3510.
- A WHO Classification-Based Prognostic Scoring System (WPSS) for Predicting Survival in Myelodysplastic Syndromes. Luca Malcovati et al. ASH Annual Meeting Abstracts 2005 106: Abstract 788.
- Scott BL, Sandmaier BM, Storer B et al. Myeloablative vs nonmyeloablative allogeneic transplantation for patients with myelodysplastic syndrome or acute myelogenous leukemia with multilineage dysplasia: a retrospective analysis. Leukemia 2006;20:128—135.
- Wallen H, Gooley TA, Deeg HJ et al. Ablative allogeneic hematopoietic cell transplantation in adults 60 years of age and older. J Clin Oncol 2005;23:3439—3446.
- Demuynck H, Verhoef GE, Zachee P et al. Treatment of patients with MDS with allogeneic bone marrow transplantation from genotypically similar HLA-identical sibling and alternative donors. Bone Marrow Transplant 1996;17:745—751.
- Bowen D. Is traditional low dose chemotherapy (cytarabine/melphalan) still on option? // Leukemia Research, Volume 31, Supplement 1, May 2007, Page S19
- Miller K.B. et al. The evaluation of low-dose cytarabine in the treatment of myelodisplastic syndromes: a phase III intergroup study. Annals of hematology, 1992; 65: 162—168.
- Burnett A.K., Milligan D., Prentice A.G. et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007. 109: 1114—1124