Байесовский подход в филогенетике
Байесовский подход в филогенетике позволяет получить наиболее вероятное филогенетическое дерево при заданных исходных данных, последовательностях ДНК или белков рассматриваемых организмов и эволюционной модели замен[1]. Для снижения вычислительной сложности алгоритма расчёт апостериорной вероятности реализуется различными алгоритмами, использующими метод Монте-Карло для марковских цепей[2]. Главными преимуществами байесовского подхода по сравнению с методами максимального правдоподобия и максимальной экономии является вычислительная эффективность, способность работать со сложными моделями эволюции, а также то, что, в отличие от методов, указывающих на единственное наилучшее по заданному критерию дерево, он позволяет выбрать несколько вариантов филогенетического дерева с наибольшим значением апостериорной вероятности[3].
История и основы метода
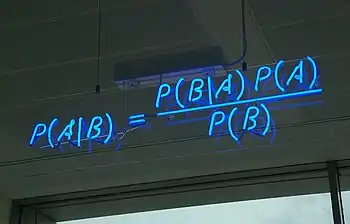
Байесовский подход является развитием вероятностного метода, разработанного английским математиком и священником Томасом Байесом на основе теоремы Байеса. Этот метод был опубликован в 1763 году[4], через два года после его смерти. Позднее современную формулировку теоремы вывел Пьер-Симон Лаплас[1] .
В 1953 году Николас Метрополис ввёл методы Монте-Карло для марковских цепей (MCMC, Markov chain Monte Carlo)[5]. Преимущества в скорости вычислений и возможность интеграции с методами MCMC позволили байесовскому подходу стать одним из самых популярных методов статистического вывода. Байесовский подход имеет множество применений в молекулярной филогенетике и систематике. По сравнению с другими методами построения филогенетических деревьев (метод максимальной экономии, метод максимального правдоподобия), он позволяет учитывать филогенетическую неопределенность, использовать априорную информацию и сложные модели эволюции, для которых традиционные методы имеют вычислительные ограничения.
Применение байесовского подхода в филогенетике состоит в следующем. Всё множество допустимых филогенетических деревьев описывается дискретными параметрами (топология деревьев) и непрерывными параметрами (длины ветвей деревьев и параметры эволюционной модели замен). Чтобы вычислить значение апостериорной плотности распределения вероятностей для дерева c топологией и параметрами , при заданных исходных данных , применяется формула Байеса , где — условная плотность распределения вероятностей исходных данных . Знаменатель в этой формуле вычисляется по формуле полной вероятности в виде суммы по интегралов от произведения по , где — априорная плотность распределения для деревьев[6]. Явные аналитическое расчеты по этой формуле не всегда возможны, а численные — требуют большого количества вычислений при поиске максимума функции по . Применение метода статистических испытаний (который также называется методом Монте-Карло) на цепях Маркова позволяет получить приближенные значения апостериорных вероятностей и уменьшить вычислительную сложность алгоритма поиска наиболее вероятного дерева по критерию максимума апостериорной вероятности.











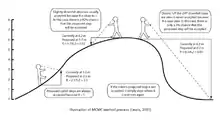
В методах MCMC апостериорная плотность вычисляется за счет имитации работы цепи Маркова, состояниями которой являются филогенетические деревья[2]. Расчет апостериорной плотности выполняется как частота посещения этих состояний в установившемся режиме. Наиболее вероятное дерево определяется по максимальной частоте того состояния, которое чаще всего посещается, или нескольких из них наиболее часто посещаемых. Методы MCMC можно описать двумя этапами: на первом применяется стохастический механизм для получения нового состояния цепи Маркова; на втором выполняется расчет вероятности перехода в это состояние и разыгрывается случайное событие смены состояния. Эта процедура повторяется тысячи или миллионы раз. Доля времени, в течение которого одиночное дерево посещается в процессе работы цепи Маркова, является достаточно точной аппроксимацией для его апостериорной вероятности. К числу наиболее часто применяемых алгоритмов, использующихся в методах MCMC, относятся алгоритм Метрополиса — Гастингса, алгоритм Метрополиса в сочетании с MCMC (MC³) и алгоритм LOCAL Ларгета и Симона.
Алгоритм Метрополиса — Гастингса
Алгоритм Метрополиcа — Гастингса[7] является одним из наиболее распространенных методов MCMC и представляет собой модифицированную Гастингсом версию алгоритма Метрополиса[5]. Алгоритм Меторополиса — Гастингса строит случайную реализацию цепи Маркова, состояниями которой являются филогенетические деревья. При имитации изменения состояния на каждом шаге выполняется переход от одного дерева к другому за счет изменения топологии или параметров эволюционной модели по определённому правилу. Алгоритм состоит из следующих шагов[8]:
- выбирается стартовое дерево со случайной топологией и параметрами модели и принимается как текущее;
- строится дерево с новой топологией или новыми параметрами модели;
- вычисляется отношение вероятностей (или функций плотности вероятности) нового дерева и старого дерева :



(под подразумевается условная вероятность или плотность распределения при заданных исходных данных );

- если , то в качестве текущего дерева принимается новое дерево ;
- если , то выбор дерева происходит с вероятностью (для этого генерируется случайное равномерно распределенное число в интервале , и если это число меньше , то в качестве текущего дерева принимается , иначе остается ;




- далее процесс повторяется с шага 2) n раз до тех пор, пока не достигнет равновесного распределения.
В оригинальном алгоритме Метрополиса предполагается, что вероятности переходов от дерева к дереву и обратно равны. Если это условие не выполняется, то применяются поправки Гастингса, состоящие в следующем: вероятность перехода вычисляется по формуле , где — совместная функция распределения.


Алгоритм Метрополиса в сочетании с МСМС
Алгоритм Метрополиса в сочетании с MCMC (Metropolis-coupled MCMC, MC³)[9], известный также как алгоритм параллельного отжига, является модифицированной версией алгоритма Метрополиса — Гастингса для марковских цепей со сложным и многомодальным распределением вероятностей состояний. Для этих случаев алгоритмы эвристического поиска деревьев критериями MP (метод максимальной экономии, maximum parsimony), ML (метод максимального правдоподобия) и ME (метод минимальной эволюции), а также МСМС могут выйти на локальный максимум, что приведёт к неверной аппроксимации апостериорной плотности распределения вероятностей. Алгоритм MC³ за счёт смешивания марковских цепей с разной температурой позволяет правильно аппроксимировать распределение апостериорных вероятностей и избегать попадания в локальные оптимумы.
Алгоритм параллельно запускает цепей, по итераций в каждой цепи с разными стационарными распределениями , , где первое распределение с целевой плотностью называется холодной цепью, а другие цепи с распределениями , называются разогретыми[10]. Плотности распределений разогретых цепей имеют вид:







- где — температурный фактор.


Возведение плотности в степень при имеет эффект уплощения распределения по аналогии с нагреванием металла. В таком распределении легче перемещаться между пиками, разделёнными долинами, чем в первоначальном распределении. После каждой итерации алгоритм предписывает выполнить обмен состояний между двумя случайно выбранными цепями с помощью предложенного Метрополисом шага. Обмен между состояниями и происходит с вероятностью:





- где — текущее состояние в цепи с номером , [11].


Эвристически, разогретые цепи будут посещать локальные пики довольно легко, и обмен состояниями между цепями позволит холодной цепи иногда перепрыгивать через долины. Если слишком мало, обмен состояниями будет редко выполняться, поэтому в алгоритме используются несколько цепей с разными температурными факторами для улучшения смешивания[6].

Для получения стационарного распределения вероятностей используются только состояния из холодной цепи, а состояния из разогретых цепей отбрасываются.
Алгоритмы GLOBAL и LOCAL
Для генерации нового состояния марковской цепи существуют различные вероятностные способы модификации деревьев, например, бисекция с последующим переприсоединением, обмен ветвей, замена на ближнее соседнее дерево. Алгоритмы LOCAL[2] и GLOBAL[12] предлагают другой способ построения нового дерева по текущему за счёт изменения топологии и длин ветвей. Это приводит к значительному сокращению вычислений для больших деревьев по сравнению с алгоритмами бутстрэпа для методов максимального правдоподобия и максимальной экономии.
Общая идея заключается в том, что дерево представляется в виде следующих параметров: топология дерева и длина его ветвей, а также параметры модели замен. При изменении состояний марковской цепи выполняются последовательные шаги, в которых отдельно либо меняется топология дерева и длина его ветвей, либо меняются только параметры модели замен. Решение о переходе к новому дереву в качестве текущего состояния цепи Маркова принимается так же, как в алгоритме Метрополиса — Гастингса, но значение пороговой вероятности вычисляется с использованием параметров модифицированного дерева.
В алгоритме GLOBAL[12], представленном Мау, Ньютоном и Ларгетом в 1999 году, все длины ветвей дерева изменяются на небольшую величину в каждом цикле. Алгоритм Ларгета и Симона LOCAL[2] предполагает модификацию дерева в небольшой окрестности случайно выбранной внутренней ветви дерева.
Построение нового дерева в алгоритме LOCAL при модификации топологии и длин ветвей выполняется по следующему правилу: равновероятно выбирается произвольное внутреннее ребро дерева с вершинами и . Вследствие того, что филогенетическое дерево должно быть бинарным, а ребро внутреннее, у каждой из вершин обязательно есть две смежные. Смежные вершины для произвольным образом обозначаются буквами и , а смежные вершины для — буквами и . Далее для вершин и равновероятно выбирается по одной смежной, например, и , и рассматривается путь между вершинами и , состоящий из трёх рёбер. Длины этих рёбер модифицируются пропорционально путём умножения на случайное число по правилу , где — старая длина пути, — новая длина пути, — это равномерно распредёленная случайная величина на отрезке , а — это положительный настраиваемый параметр. Следующий шаг модификации дерева состоит в отсоединении одной из вершин, или , выбираемых равновероятно, и присоединения её в случайно выбранной по равномерному закону точке на пути от вершины до вершины вместе с её дочерней ветвью. При такой модификации возможно изменение топологии дерева, если порядок следования вершин и вдоль пути от к изменился, иначе — топология дерева не изменяется. Поправка Гастингса равна квадрату отношения длин нового и старого пути: .











При модификации параметров модели в алгоритме рассматриваются два варианта: в первом варианте, когда один параметр ограничен набором значений , новое значение параметра вычисляется прибавлением равномерно распределённой случайной величины из интервала . Если новое значение выходит за пределы допустимого диапазона [2], то остаток отражается внутрь этого отрезка. Поправка Гастингса принимается равной 1. Второй вариант составляет случай, когда модифицируется множество параметров, сумма которых равна константе. В этом случае новое множество значений этих параметров выбирается из распределения Дирихле, центрированного по текущим значениям параметров. Поправка Гастингса вычисляется как отношение плотностей Дирихле с новыми и старыми параметрами.


Критика и обсуждение
- Значения бутстрэп против апостериорных вероятностей. Было показано, что значения бутстрэпа, вычисленные методами максимальной экономии или правдоподобия, обычно ниже, чем апостериорные вероятности, полученные байесовским методом[13]. Это приводит ряду вопросов, например: Приводят ли апостериорные вероятности к завышению вероятности результата? Являются ли значения бутстрэпа более устойчивыми по сравнению с апостериорными вероятностями?
- Выбор модели. Результаты Байесовского филогенетического анализа прямо коррелируют с выбранной моделью эволюции, поэтому важно выбрать модель, которая подходит наблюдаемым данным, иначе вывод о филогении может быть ошибочным. Многие ученые поднимали вопрос об интерпретации результатов байесовского анализа, когда модель неизвестна или неправильная. Например, слишком упрощенная модель может дать более высокие апостериорные вероятности[14][15] или простые модели связаны с большей неопределённостью, чем с той, которая вытекает из анализа бустрэпом.
Программа MRBAYES
MrBayes — это бесплатная программа, осуществляющая байесовский анализ филогении. Первоначально написана Джоном Хюльсенбеком и Фредериком Ронкустом в 2001 году[16]. Когда байесовские методы приобрели популярность, многие молекулярные филогенетики стали выбирать MrBayes. Программа использует стандартный алгоритм MCMC и алгоритм Метрополиса, связанный с MCMC.
MrBayes использует МСМС для приближённого вычисления апостериорных вероятностей деревьев[5]. Пользователь может поменять предположения о модели замен, априорных вероятностей и деталей МС анализа. Также программа позволяет удалять и добавлять таксоны и символы для анализа. В программе можно использовать широкий спектр моделей замен -- от стандартной модели подстановок DNA 4х4, также называемой JC69, в которой считается, что частоты оснований равны и все замены нуклеотидов происходят с равной вероятностью[17], до наиболее общей модели GTR, в которой различаются и частоты оснований, и вероятности замен. Также программа включает несколько 20х20 моделей замен аминокислот, кодоновые и дублетные модели замены ДНК. Программа предлагает различные методы для ослабления предположения о равных скоростях замен в нуклеотидных позициях[18]. MrBayes также может выводить наследственные состояния, содержащие неопределённость филогенетического дерева и параметров модели.
MrBayes 3[19] — это полностью реогранизованная и реконструированная версия первоначальной программы MrBayes. Главное новшество заключается в возможности программы приспосабливаться к неоднородности наборов данных. Такая структура позволяет пользователю смешивать модели и получать преимущество эффективности байесовского MCMC анализа, если он имеет дело с разными типами данных (например, белки, нуклеотиды, морфологические данные). По умолчанию программа использует алгоритм МСМС Метрополиса.
MrBayes 3.2 — это новая версия MrBayes, выпущенная в 2012 году[20]. Новая версия позволяет пользователю запускать несколько анализов параллельно. Также она обеспечивает более быстрое вычисление вероятностей и даёт возможность использования ресурсов графического процессора для выполнения этих вычислений. Версия 3.2 предоставляет больше опций для выходных данных, совместимых с программой FigTree и другими программами для просмотра деревьев.
Список программ, использующих байесовский подход
| Название программы | Описание | Метод | Авторы | Ссылка на сайт |
|---|---|---|---|---|
| Armadillo Workflow Platform | Программа, предназначенная для филогенетического и общего биоинформатического анализа | Вывод филогенетических деревьев с использованием методов ML, MP, байесовскийого подхода и др. | E. Lord, M. Leclercq, A. Boc, A.B. Diallo, V. Makarenkov[21] | https://web.archive.org/web/20161024081942/http://www.bioinfo.uqam.ca/armadillo/. |
| Bali-Phy | Одновременное получение выравнивания и дерева на основе байесовскийого похода | Байесовский вывод выравниваний и филогенетических деревьев | M.A. Suchard, B. D. Redelings[22] | http://www.bali-phy.org |
| BATWING | Вывод деревьев методом Байеса с созданием внутренних узлов | Байесовский анализ, демографическая история, метод расщепления популяций | I. J. Wilson, D. Weale, D.Balding[23] | http://heidi.chnebu.ch/doku.php?id=batwing |
| Bayes Phylogenies | Вывод деревьев методом Байеса с использованием методов Монте-Карло для марковских цепей и Метрополиса в сочетании с MCMC | Байесовский анализ, множественные, смешанные модели (с автоматическим разбиением) | M. Pagel, A. Meade[24] | http://www.evolution.rdg.ac.uk/BayesPhy.html |
| PhyloBayes / PhyloBayes MPI | MCMC семплер для филогенетических реконструкций. | MCMC, вероятностная модель CAT, учитывающая сайт-специфичные нуклеотиды или аминокислоты | N. Lartillot, N. Rodrigue, D. Stubbs, J. Richer[25] | https://web.archive.org/web/20181218053945/http://www.phylobayes.org/ |
| BEAST | Анализ молекулярных последовательностей с помощью MCMC (Bayesian Evolutionary Analysis Sampling Trees) | Байесовский анализ, релаксированные молекулярные часы, демографическая история | A. J. Drummond, A. Rambaut & M. A. Suchard[26] | http://beast.bio.ed.ac.uk |
| BUCKy | Байесовское соответствие филогенетических деревьев для генов | Байесовское соответствие с использованием модифицированного жадного консенсуса для неукоренённых квартетов | C. Ané, B. Larget, D.A. Baum, S.D. Smith, A. Rokas, B. Larget, S.K. Kotha, C.N. Dewey, C. Ané[27] | http://www.stat.wisc.edu/~ane/bucky/ |
| Geneious (MrBayes plugin) | Средства для исследования геномов и протеомов | Neighbor-joining, UPGMA, плагины MrBayes, PHYML, RAxML, FastTree, GARLi, PAUP* | A. J. Drummond,M.Suchard,V.Lefort и др.[28] | http://www.geneious.com |
| TOPALi | Филогенетический вывод | Выбор филогенетической модели, байесовскийий анализ и оценка филогенетических деревьев методом максимального правдоподобия, определение сайтов, находящихся под позитивным отбором, анализ положения точек рекомбинации | I.Milne, D.Lindner и др.[29] | http://www.topali.org |
Приложения
Байесовский подход широко используется молекулярными филогенетиками для различных приложений:
- Вывод филогений[30][31]
- Вывод и оценка неопределенности филогений[32]
- Датирование с помощью молекулярного анализа[33]
- Вывод черт предковых форм[34][35]
- Вывод ареалов предковых форм[36]
- Модель динамики диверсификации и вымирания видов[37]
- Объяснение закономерностей распространения патогенных организмов[38]
Примечания
- Laplace, P. Memoir on the Probability of the Causes of Events (англ.) // Statistical Science : онлайн-журнал. — 1986. — Iss. 1(3). — ISSN 359—378.
- Larget, B. & Simon, D. L. Markov Chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. (англ.) // Mol. Biol. Evol. : онлайн-журнал. — 1999. — Iss. 16. — ISSN 750–759.
- Mark Holder1 & Paul O. Lewis1. Phylogeny estimation: traditional and Bayesian approaches (англ.) // Nature Reviews Genetics : онлайн-журнал. — 2003. — Iss. 4. — ISSN 275-284. — doi:10.1038/nrg1044.
- Bayes Thomas ,Price Richard. An Essay towards solving a Problem in the Doctrine of Chance. By the late Rev. Mr. Bayes, communicated by Mr. Price, in a letter to John Canton, A. M. F. R. S. (англ.) // Philosophical Transactions of the Royal Society of London : онлайн-журнал. — 1763. — Iss. 53. — doi:10.1098/rstl.1763.0053. Архивировано 10 апреля 2011 года.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, E. Teller. [http://aip.scitation.org/doi/abs/10.1063/1.1699114 Equation of state calculations by fast computing machines] (англ.) // The Journal of Chemical Physics : онлайн-журнал. — Macmillan Publishers Limited, 1953. — Iss. 21. — ISSN 1087-1092. — doi:10.1063/1.1699114.
- Rannala B., Yang Z. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. (англ.) // J. Mol. Evol. : онлайн-журнал. — 1996. — Iss. 43. — ISSN 304–311.
- Hastings,W.K. Monte Carlo sampling methods using Markov chains and their applications (англ.) // Biometrika : онлайн-журнал. — Oxford Academic, 1970. — Iss. 57. — ISSN 97-109.
- Felsenstein, J. Inferring phylogenies. — Massachusetts: Sinauer Associates, 2004. — 664 с.
- Geyer,C.J. Markov chain Monte Carlo maximum likelihood. (англ.) // Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface. : онлайн-журнал. — Interface Foundation, Fairfax Station, 1991.
- Li, S., Pearl, D. K. & Doss, H. Phylogenetic tree construction using Markov Chain Monte Carlo. (англ.) // J. Am. Stat. Assoc. : онлайн-журнал. — 2000. — Iss. 95. — ISSN 493–508.
- Yang, Z. H. & Rannala, B. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo method. (англ.) // Mol. Biol. Evol. : онлайн-журнал. — 1997. — Iss. 14. — ISSN 717–724.
- Mau,B., Newton,M. and Larget,B. Bayesian phylogenetic inference via Markov chain Monte carlo methods. (англ.) // Biometrics : онлайн-журнал. — 1999. — Iss. 55(1). — ISSN 1-12.
- Garcia-Sandoval, R. Why some clades have low bootstrap frequencies and high Bayesian posterior probabilities. (англ.) // Israel Journal of Ecology & Evolution : онлайн-журнал. — 2014. — Iss. 60(1). — ISSN 41-44.
- Suzuki, Y. et al. Over credibility of molecular phylogenies obtained by Bayesian phylogenetics. (англ.) // Proc. Natl. Acad. Sci. U. S. A. : онлайн-журнал. — 2002. — Iss. 99. — ISSN 16138-16143.
- Erixon, P. et al. Reliability of Bayesian posterior probabilities and bootstrap frequencies in phylogenetics. (англ.) // Syst. Biol. : онлайн-журнал. — 2003. — Iss. 52. — ISSN 665—673.
- Huelsenbeck, J. P. and F. Ronquist. MrBayes: Bayesian inference of phylogeny. (англ.) // Bioinformatics : онлайн-журнал. — 2001. — Iss. 17. — ISSN 754-755.
- Jukes, T.H. and Cantor, C.R. Evolution of Protein Molecules. (англ.) // New York: Academic Press : онлайн-журнал. — 1969.
- Yang, Z. Maximum likelihood estimation of phylogeny from DNA sequences when substitutions rates differ over sites. (англ.) // Mol. Biol. Evol. : онлайн-журнал. — 1993. — Iss. 10. — ISSN 1396—1401.
- Ronquist F., Huelsenbeck J.P. Mrbayes 3: Bayesian phylogenetic inference under mixed models. (англ.) // Bioinformatics : онлайн-журнал. — 2003. — Iss. 19. — ISSN 1572-1.
- Ronquist F., TeslenkoM.,Van Der Mark P.,Ayres D.L., DarlingA.,Hhna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. (англ.) // Syst. Biol. : онлайн-журнал. — 2012. — Iss. 61. — ISSN 539-542.
- Lord, E., Leclercq, M., Boc, A., Diallo, A.B., Makarenkov, V. Armadillo 1.1: An Original Workflow Platform for Designing and Conducting Phylogenetic Analysis and Simulations. (англ.) // PLoS ONE : онлайн-журнал. — 2012. — Iss. 7(1). — doi:10.1371/journal.pone.0029903.
- Suchard, M.A. and Redelings, B.D. BAli-Phy: simultaneous Bayesian inference of alignment and phylogeny (англ.) // Bioinformatics : онлайн-журнал. — 2006. — Iss. 22. — ISSN 2047-2048.
- Wilson, I., Weale, D. and Balding, M. Inferences from DNA data: population histories, evolutionary processes and forensic match probabilities. (англ.) // Journal of the Royal Statistical Society: Series A (Statistics in Society) : онлайн-журнал. — 2003. — Iss. 166. — ISSN 155—188.
- Pagel, M. and Meade, A. Bayesian analysis of correlated evolution of discrete characters by reversible-jump Markov chain Monte Carlo. (англ.) // American Naturalist : онлайн-журнал. — 2006. — Iss. 167. — ISSN 808—825.
- Lartillot N., Philippe H. A Bayesian Mixture Model for Across-Site Heterogeneities in the Amino-Acid Replacement Process. (англ.) // Molecular Biology and Evolution : онлайн-журнал. — 2004. — Iss. 21(6). — ISSN 1095-1109.
- Drummond, A.J., Rambaut, A. Beast: Bayesian evolutionary analysis by sampling trees. (англ.) // BMC Evol Biol. : онлайн-журнал. — 2007. — Iss. 7:214.
- Ané, C., Larget, B., Baum, D.A.,Smith, S.D., Rokas, A. Bayesian estimation of concordance among gene trees. (англ.) // Molecular Biology and Evolution : онлайн-журнал. — 2007. — Iss. 24(2). — ISSN 412—426.
- Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Mentjies, P., & Drummond, A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. (англ.) // Bioinformatics : онлайн-журнал. — 2012. — Iss. 28(12). — ISSN 1647-1649.
- Milne, I., Lindner, D., Bayer, M., Husmeier, D., McGuire, G., Marshall, D.F. and Wright, F. TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. (англ.) // Bioinformatics : онлайн-журнал. — 2008. — Iss. 25(1). — ISSN 126—127.
- Alonso, R., Crawford, A.J. & Bermingham, E. Molecular phylogeny of an endemic radiation of Cuban toads (Bufonidae: Peltophryne) based on mitochondrial and nuclear genes. (англ.) // Journal of Biogeography : онлайн-журнал. — 2011. — Iss. 39 (3). — ISSN 434—451.
- Antonelli, A., Sanmart.n, I. Mass Extinction, Gradual Cooling, or Rapid Radiation? Reconstructing the Spatiotemporal Evolution of the Ancient Angiosperm Genus Hedyosmum (Chloranthaceae) Using Empirical and Simulated Approaches. (англ.) // Syst. Biol. : онлайн-журнал. — 2011. — Iss. 60(5). — ISSN 596-615.
- Bacon, C.D., Baker, W.J., Simmons, M.P. Miocene dispersal drives island radiations in the palm tribe Trachycarpeae (Arecaceae). (англ.) // Systematic Biology : онлайн-журнал. — 2012. — Iss. 61. — ISSN 426—442.
- Särkinen, T., Bohs, L., Olmstead,R.G. and Knapp, S. A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. (англ.) // BMC Evolutionary Biology. : онлайн-журнал. — 2013.
- Ronquist, F. Bayesian inference of character Evolution. (англ.) // Trends in Ecology and Evolution : онлайн-журнал. — 2004. — Iss. 19 No.9. — ISSN 475—481.
- Schäffer , S., Koblmüller, S., Pfingstl, T., Sturmbauer, C., Krisper, G. Ancestral state reconstruction reveals multiple independent evolution of diagnostic morphological characters in the «Higher Oribatida» (Acari), conflicting with current classification schemes. (англ.) // BMC Evolutionary Biology : онлайн-журнал. — 2010. — Iss. 10:246.
- Filipowicz, N., Renner, S. Brunfelsia (Solanaceae): A genus evenly divided between South America and radiations on Cuba and other Antillean islands. (англ.) // Molecular Phylogenetics and Evolution : онлайн-журнал. — 2012. — Iss. 64. — ISSN 1-11.
- Silvestro, D., Schnitzler, J., Liow, L.H., Antonelli, A., Salamin, N. Bayesian Estimation of Speciation and Extinction from Incomplete Fossil Occurrence Data. (англ.) // Syst. Biol. : онлайн-журнал. — 2014. — Iss. 63(3). — ISSN 349-367.
- Lemey, P., Rambaut, A., Drummond, A.J., Suchard, M.A. Bayesian Phylogeography Finds Its Roots. (англ.) // PLoS Comput Biol : онлайн-журнал. — 2009. — Iss. 5(9).