Синдром Аперта
Синдром Апе́ра (Аперта) (акрокраниодисфалангия, акросфеносиндактилия, акроцефалосиндактилия) — врожденная аномалия развития черепа, которая сочетается с отклонением развития кистей рук. Раннее закрытие венечного и стреловидного швов способствует деформации черепа, что приводит к внутричерепной гипертензии[1]. Синдром Аперта является одной из форм акроцефалосиндактилии[прим. 1].
| Синдром Аперта | |
|---|---|
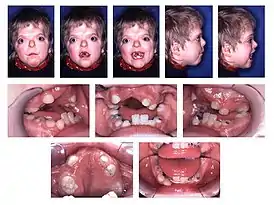 Дети с синдромом Аперта | |
| МКБ-11 | LD24.G2 |
| МКБ-10 | Q87.0 |
| МКБ-9 | 755.55 |
| OMIM | 101200 |
| DiseasesDB | 33968 |
| MedlinePlus | 001581 |
| eMedicine | ped/122 |
| MeSH | D000168 |
Впервые это заболевание описал французский педиатр Эжен Аперт в 1906 году[2]. Заболевание встречается у одного новорожденного на 160 000—200 000[3].
Этиология
Акрокраниодисфалангия — заболевание с аутосомно-доминантным типом наследования[прим. 2]. У детей, родители которых страдают синдромом Аперта, вероятность унаследовать заболевание равна 50%. В 1995 году были опубликованы доказательства, судя по которым, причиной возникновения данного синдрома является нарушение работы генов 10-й хромосомы человека[4].
Признаки и симптомы

Одним из признаков заболевания является то, что краниосиностоз[прим. 3] сочетается с брахикефалией. В связи с преждевременным срастанием коронарных швов увеличивается внутричерепное давление, что обычно приводит к умственной отсталости. Немаловажными признаками синдрома являются высокий, выпуклый лоб, плоское или вогнутое лицо, в результате чего наблюдается нарушение костей лицевого черепа, что приводит к деформации челюсти, а также синдактилия рук и ног с вовлечением 2, 3 и 4-го пальцев.
Лечение
Радикальных методов лечения не существует.
Симптоматическое лечение данной патологии заключается в хирургическом увеличении объёма черепа, коррекции синдактилии и полидактилии.
Хирургическое лечение включает в себя раннюю краниоэктомию коронарного шва и фронто-орбитальную репозицию для уменьшения проявлений дисморфизма и патологических изменений формы черепа. Операции по поводу синдрома Аперта часто состоят из нескольких этапов, последний проводится в подростковом возрасте. Первый этап часто выполняется уже в 3 мес.[5]
Консервативные методы лечения применяют для разработки суставов. Также, для стимуляции психического развития назначают ноотропные препараты: аминалон, пирацетам и другие[1], использовались в эпоху развития науки до проверки лечения методами доказательной медицины.
Примечания
- Акроцефалосиндактилия — группа наследственных пороков развития черепа и пальцев.
- Аутосомно-доминантный тип наследования — тип наследования, при котором мутантный аллель доминирует над нормальным аллелем, в связи с чем болезнь или признаки болезни ярко выражены.
- Краниосиностоз — преждевременное сращение некоторых костей черепа.
Источники
- Л.О. Бадалян. Детская неврология. — Москва: Медицина, 1984. — С. 347—349. — 576 с.
- Apert's syndrome (whonamedit.com) (англ.). Дата обращения: 13 августа 2010. Архивировано 6 мая 2012 года.
- Kaplan, L C. Clinical assessment and multispecialty management of Apert syndrome (англ.) // Clinics in plastic surgery : journal. — 1991. — April (vol. 18, no. 2). — P. 217—225. — ISSN 00941298. — PMID 2065483.
- Wilkie, A O; S. F. Slaney, M. Oldridge, M. D. Poole, G. J. Ashworth, A. D. Hockley, R. D. Hayward, D. J. David, L. J. Pulleyn, P. Rutland. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome (англ.) // Nature genetics : journal. — 1995. — February (vol. 9, no. 2). — P. 165—172. — doi:10.1038/ng0295-165. — PMID 7719344.
- Синдром Апера: клинические проявления и этиология — Интернет-сообщество нейрохирургов Росcии. neuro-online.ru. Дата обращения: 4 марта 2017.
