Клеточное старение
Клеточное старение — явление, которое обычно связывают с потерей способности клетки к делению (предел Хейфлика). Этот процесс также называется репликативным старением. В русскоязычной литературе под термином клеточное старение ещё понимают снижение функциональной активности клеток по мере увеличения их возраста.
Клетки, растущие в культуре, могут делиться только определённое число раз, после чего переходят в стадию старения. Такие клетки характеризуются снижением интенсивности энергообмена, замедлением синтеза РНК и белков, понижением эффективности репарации ДНК и накоплением мутаций. Часто наблюдается разбалансировка клеточной регуляции. Признаками клеточного старения считаются накопление специфического гликолипопротеида липофусцина и активация бета-галактозидазы. Клеточное старение было освещено на клетках разного типа[1].
Стареющие клетки могут долгое время оставаться жизнеспособными. Нередко после остановки деления и торможения клеточного цикла у них не наступает программируемой клеточной гибели. Обычно они уничтожаются клетками иммунной системы. С возрастом в организме происходит накопление старых клеток, вероятно, вследствие ухудшения выполнения иммунной системой своих функций.
Стареющие клетки могут оказывать влияние как на соседние клетки, так и на весь организм, выделяя определённые сигнальные молекулы. Влияние это разнообразно, изучено недостаточно и, в общем, скорее отрицательно[2]. Похоже, что клеточное старение является одним из механизмов старения организма.
Механизм клеточного старения
Укорочение теломер
Укорочение теломер, концевых участков ДНК на концах хромосом — одна из основных причин ограничения числа клеточного деления и клеточного старения. Функцией теломер являются защита хромосом от деградации и «слипания» их друг с другом. Анализ длины теломерных повторов выявил, что соматические клетки теряют от 50 до 200 нуклеотидов при каждом клеточном делении[3]. Это происходит из-за того, что ДНК-полимераза не способна реплицировать концы молекул ДНК. При отсутствии в клетках активной теломеразы после определённого числа делений происходит сильное укорочение теломер, и клетка перестает делиться. Число клеточного деления, которое произошло до этого момента, носит название предела Хейфлика. Для большинства соматических клеток человека этот предел составляет около пятидесяти фаз деления.
Сама идея счета фаз деления и старения вследствие недорепликации ДНК на концах хромосом (теломерных участков) принадлежит российскому учёному А. М. Оловникову. Теория была выдвинута в 1971 году для объяснения экспериментальных данных Леонарда Хейфлика и получила название маргинотомии.
Считается, что укорочение хромосом до определённого размера обусловливает процесс клеточного старения, а длина теломер, по этим представлениям, может служить мерой клеточного потенциала деления[4].
Следует отметить, что у клеток больных синдромом Хатчинсона — Гилфорда (детская прогерия) предел Хейфлика значительно снижен. Схожая картина наблюдается у больных синдромом Вернера (прогерия взрослых). В этом случае больные нормально доживают до 17-18 лет, но начинают стремительно стареть, перейдя этот рубеж. Теломеры у таких больных нормальной длины, но из-за мутаций их ДНК более чувствительна к разрушению, чем ДНК здорового человека.
Согласно ещё одной модели, у молодых клеток пока ещё длинные теломеры находятся в области гетерохроматина. По мере укорочения теломер область гетерохроматина включает в себя всё больше субтеломерной ДНК, где, возможно, находится некий ген-супрессор, подавляющий программу клеточного старения. Инактивация этого гена путём включения его в область гетерохроматина и приводит к запуску процесса старения[5].
Роль фосфоинозитид-3-киназы
Фосфоинозитид-3-киназа (PI3K) контролирует пролиферацию клеток и апоптоз. Существуют данные о влиянии PI3K на регуляцию клеточного старения.
Ген Age1 нематоды Caenorhabditis elegans является гомологом гена млекопитающих, кодирующего каталитическую субъединицу PI3K. Мутации в гене Age1 значительно увеличивают продолжительность жизни червей[6].
Ингибирование PI3K в культуре человеческих фибробластов приводит к торможению их пролиферации. Клетки демонстрируют признаки, характерные для стареющих клеток: активацию бета-галактозидазы, повышение экспрессии гена коллагеназы и подавление экспрессии специфического маркера пролиферирующих фибробластов, гена EPC-1 (англ. early population doubling level cDNA 1)[7].
Признаки клеточного старения
Изменение ответа на факторы роста
По мере старения клеток уменьшается их способность реагировать на определённые внешние стимулы. Эффект действия факторов роста, гормонов и других стимулирующих агентов на старые клетки гораздо ниже, чем на молодые, способные к активному делению. Токсины, антибиотики, радиация и тепловой шок, напротив, оказывают на них более сильное воздействие.
Известно, что культура клеток пациентов, страдающих синдромом преждевременного старения, таким как прогерия и синдром Вернера, дает значительно более низкий ответ на стимуляцию инсулином, сывороткой и другими факторами, чем клетки здоровых людей[8].
Рецепторная система клеток существенно не меняются при старении. Таким образом, снижение клеточного ответа на фактор роста не связано с уменьшением количества их рецепторов.
Остановка клеточного цикла
При старении клеток наблюдается необратимая блокировка клеточного цикла. Точный механизм, мешающий клетке перейти в S-фазу, пока неизвестен. Однако отмечается, что при пролиферативном старении клеток отсутствует экспрессия некоторых генов, обеспечивающих протекание клеточного цикла. В стареющих клетках подавлена экспрессия циклинов, Cdk2[9], инсулиноподобного фактора роста 1 (IGF-1)[10], а также некоторых других факторов. При этом никакие экзогенные факторы, в том числе IGF-1, не могут вывести «старую» клетку из состояния неспособности к делению.
Существует мнение, что апоптоз и переход клеток в стадию покоя являются альтернативной защитной реакцией на действие повреждающих агентов и необходимы для профилактики онкогенной трансформации клеток. Если повреждённая клетка по той или иной причине не переходит к апоптозу или клеточному старению, она может стать злокачественной[11].
Клеточное старение и рак
Болезни, связанные со старостью разделяются на две большие категории. Первую группу составляют болезни, связанные с утратой функции, в основном, это дегенеративные заболевания (например, болезнь Альцгеймера, болезнь Паркинсона, саркопения, макулодистрофия и т. д.). Вторая группа состоит из болезней, связанных с усилением функции (аденома простаты,атеросклероз и другие). Наиболее известной и смертоносной из них является рак. Фактором риска для образования злокачественной опухоли являются влияние генетических факторов и окружающей среды, но наиболее значимый из них это фактор зрелого возраста. Вероятность опухолеобразования после 50 лет возрастает почти экспоненциально. Во-первых, это вызвано тем, что с возрастом накапливаются мутации, способствующие онкогенезу. Доказательством этого служит, например, что у людей с мутациями в генах, вызывающими рак, образование опухоли происходит в раннем возрасте. Также генетическая нестабильность (дестабилизация хромосом, обмен сестринских хроматид, анеуплоидия, мутации и амплификации генов, клональная гетерогенность, неопластическая трансформация) может влиять на онкогенез. Во-вторых, накопление стареющих клеток образует среду благоприятную для опухолеобразования. Нормальное тканевое микроокружение может подавить способность мутировавших раковых клеток к размножению и выживанию, поэтому опухолевые клетки часто должны уметь модифицировать окружающую тканевую среду. Однако тканевое микроокружение само может обладать проканцерогенным состоянием независимо от присутствия раковых клеток. Возникновению такого состояния может способствовать возраст. Механизм с помощью которого возраст вызывает состояние, благоприятствующее онкогенезу, является мультифакторным и, до конца, неизученным. Одним из таких факторов является клеточное старение. Например, повреждение или стресс, подвергающий пролиферирующую клетку риску злокачественной трансформации, вызывает клеточное старение, защищая клетки от рака. Это связано с работой p53 и p16INK4a/pRB, являющегося наиболее значимым противоопухолевым механизмом. Следовательно, для онкогенеза необходима генетическая (мутагенная) или эпигенетическая инактивация этого эффективного механизма[12].
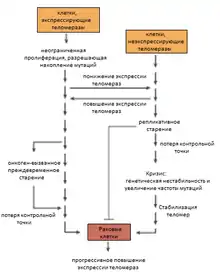
Опухоль может образоваться как из клеток, которые экспрессируют теломеразы (например, стволовые клетки), так и из клеток, которые этого не делают. В клетках, в которых теломеразы отсутствуют, укорочение теломер может вызвать репликативное старение, которое предназначено предотвращать развитие рака. И, наоборот, короткие теломеры могут привести к увеличению генетической нестабильности и, соответственно, к образованию опухоли. А в клетках экспрессирующих теломеразы, её выключение может вызвать генетическую нестабильность[13].
Однако клеточное старение может вызвать и развитие рака. Сначала эта идея кажется парадоксальной, но эволюционная теория антагонистической плейотропии предусматривает, что биологический процесс может быть как полезным, так и вредным, в зависимости от возраста. Большинство животных развивается в условиях, изобилующих смертельной внешней опасностью (хищники, инфекции, голод и т. д.). В этих условиях пожилая особь представляет собой редкость и поэтому отбор против процессов, которые на поздних этапах жизни способствуют возникновению болезни, является слабым. То есть, они избегают влияния процесса естественного отбора. Таким образом, биологический процесс, который был нужен для развития выносливости у молодого организма (например, подавление опухолеообразования) может нести вред для зрелого организма, (вызывая болезни позднего возраста, включая рак)[14].
Литература
- Sheloukhova L. (2019). Сенесценция RLEgroup (radical life extension group) Краткий обзор по клеточному старению и разрабатываемым методам борьбы с ним с помощью сенолитиков
Примечания
- Fernandes P. B., Panos C. Wall-less microbial isolate from a human renal biopsy. (англ.) // Journal of clinical microbiology. — 1977. — Vol. 5, no. 1. — P. 106—107. — PMID 833264.
- Campisi J., d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. (англ.) // Nature reviews. Molecular cell biology. — 2007. — Vol. 8, no. 9. — P. 729—740. — doi:10.1038/nrm2233. — PMID 17667954.
- Harley C. B., Futcher A. B., Greider C. W. Telomeres shorten during ageing of human fibroblasts. (англ.) // Nature. — 1990. — Vol. 345, no. 6274. — P. 458—460. — doi:10.1038/345458a0. — PMID 2342578.
- Harley C. B., Vaziri H., Counter C. M., Allsopp R. C. The telomere hypothesis of cellular aging. (англ.) // Experimental gerontology. — 1992. — Vol. 27, no. 4. — P. 375—382. — PMID 1459213.
- Kim N. W., Piatyszek M. A., Prowse K. R., Harley C. B., West M. D., Ho P. L., Coviello G. M., Wright W. E., Weinrich S. L., Shay J. W. Specific association of human telomerase activity with immortal cells and cancer. (англ.) // Science (New York, N.Y.). — 1994. — Vol. 266, no. 5193. — P. 2011—2015. — PMID 7605428.
- Morris J. Z., Tissenbaum H. A., Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. (англ.) // Nature. — 1996. — Vol. 382, no. 6591. — P. 536—539. — doi:10.1038/382536a0. — PMID 8700226.
- Tresini M., Mawal-Dewan M., Cristofalo V. J., Sell C. A phosphatidylinositol 3-kinase inhibitor induces a senescent-like growth arrest in human diploid fibroblasts. (англ.) // Cancer research. — 1998. — Vol. 58, no. 1. — P. 1—4. — PMID 9426047.
- Bauer E. A., Silverman N., Busiek D. F., Kronberger A., Deuel T. F. Diminished response of Werner's syndrome fibroblasts to growth factors PDGF and FGF. (англ.) // Science (New York, N.Y.). — 1986. — Vol. 234, no. 4781. — P. 1240—1243. — PMID 3022382.
- Afshari C. A., Vojta P. J., Annab L. A., Futreal P. A., Willard T. B., Barrett J. C. Investigation of the role of G1/S cell cycle mediators in cellular senescence. (англ.) // Experimental cell research. — 1993. — Vol. 209, no. 2. — P. 231—237. — doi:10.1006/excr.1993.1306. — PMID 8262140.
- Ferber A., Chang C., Sell C., Ptasznik A., Cristofalo V. J., Hubbard K., Ozer H. L., Adamo M., Roberts C. T. Jr., LeRoith D. Failure of senescent human fibroblasts to express the insulin-like growth factor-1 gene. (англ.) // The Journal of biological chemistry. — 1993. — Vol. 268, no. 24. — P. 17883—17888. — PMID 7688732.
- Laura L. Mays Hoopes, Ph.D. (Dept. of Biology, Pomona College) © 2010 Nature Education Citation: Mays Hoopes, L. L. (2010) Aging and Cell Division. Nature Education 3(9):55
- Campisi J., Andersen J. K., Kapahi P., Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? (англ.) // Seminars in cancer biology. — 2011. — Vol. 21, no. 6. — P. 354—359. — doi:10.1016/j.semcancer.2011.09.001. — PMID 21925603.
- Mathon N. F., Lloyd A. C. Cell senescence and cancer. (англ.) // Nature reviews. Cancer. — 2001. — Vol. 1, no. 3. — P. 203—213. — doi:10.1038/35106045. — PMID 11902575.
- Rodier F., Campisi J. Four faces of cellular senescence. (англ.) // The Journal of cell biology. — 2011. — Vol. 192, no. 4. — P. 547—556. — doi:10.1083/jcb.201009094. — PMID 21321098.