Болезнь Галлервордена — Шпатца
Нейродегенерация с отложением железа в мозге или Болезнь Галлервордена — Шпатца — очень редкое нейродегенеративное заболевание, сопровождающееся отложением железа в базальных ганглиях (в бледном шаре и в чёрной субстанции). Это аутосомно-рециссивно наследуемое заболевание было впервые описано в 1922 году Юлиусом Галлеворденом и Гуго Шпатцем. Частота заболевания в среднем 1-3 человека на 1 миллион.
| Нейродегенерация с отложением железа в мозге или Синдром Галлервордена — Шпатца | |
|---|---|
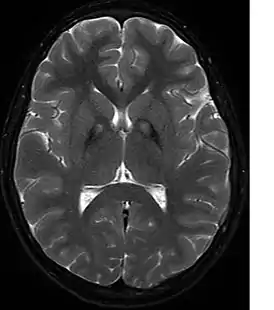 На МРТ изображены отложения железа в базальных ганглиях. | |
| МКБ-10 | G23.0 |
| МКБ-10-КМ | G23.0 |
| МКБ-9 | 333.0 |
| МКБ-9-КМ | 333.0[1] |
| OMIM | 234200 |
| DiseasesDB | 29462 |
| MedlinePlus | 001225 |
| eMedicine | neuro/151 |
| MeSH | D006211 |
Патогенез
У большинства пациентов, особенно при ранней манифестации заболевания, определяются мутации в кодируемом фермент пантотенкиназу (PANK2) гене, в хромосоме 20p13. Этот фермент играет решающую роль в биосинтезе кофермента-А. Дефект фермента ведет к накоплению цистеина, который в присутствии железа (что означает — в особенности в области чёрного вещества и базальных ганглиев) ведет к повышению свободных радикалов и способствует оксидатному повреждению мозга. Процесс в целом приводит к отложению железа и нейромеланина.
Протекание заболевания
Обычное начало заболевания в детском возрасте, иногда с выраженной симптоматикой уже на первом году жизни. Редко возможна манифестация во взрослом возрасте. Сначала возникают экстрапирамидные моторные нарушения, в особенности нарушения ходьбы с тенденцией к падениям или дистонией ног, реже — психические нарушения. В дальнейшем — нарушения движений (дистонии, хореоатетоз, тремор) с ригидностью затылочной мускулатуры, гиперрефлексией и нарушениями психики (как правило, расстройство интеллекта в виде прогрессирующей деменции). Также обнаруживаются дизартрия и дисфагия. Можно обнаружить пигментацию сетчатки или атрофию зрительного нерва[2]. У взрослых преобладает синдром паркинсонизма-плюс — с деменцией, гиперрефлексией и проминентной дистонией. Течение заболевания прогрессивно, неврологическое состояние пациентов ухудшается постепенно.
Смерть наступает обычно в возрасте до 30 лет, в состоянии полного психического распада личности.
Дополнительная диагностика
Необходимо биохимическое исключение болезни Вильсона-Коновалова, факультативно — исключение нейроакантоцитоза, прежде всего с помощью МРТ. В МРТ в Т2- взвешенных изображениях являются типичными — обусловленные отложением железа — гипоинтенсивные очаги в бледном шаре, с центральным очагом гиперинтенсивности — так называемые «глаза тигра». Этот симптом обнаруживается у всех больных с PANK2-мутациями. В генетическом обследовании могут обнаруживаться мутации в PANK2-гене. Тем не менее, уверенно можно говорить о диагнозе только после патологоанатомического исследования.
Лечение
Каузальная (этиологическая) терапия неизвестна. Были попытки лечения энзимного дефекта. Хелаторы («ловушки») железа, такие как Дефероксамин, не оказывают эффекта, тем не менее, с 2007 года проводятся попытки проводить лечение хелатором железа Феррипрокс (Деферипрон®). В экспериментах на животных глубокая стимуляция мозга приводила к усилению дистоний и гиперкинезов. Гипокинезия может лечиться Леводопой, гиперкинезы — антихолинэргиками. Тем не менее, эффект Леводопы у пациентов с мутацией гена PANK2 очень сомнителен. Для мышечной релаксации и купирования болевого синдрома часто назначается Баклофен или бензодиазепины.
Название заболевания
Из-за связей Юлиуса Галлервордена с программой эвтаназии в фашистской Германии было предложено называть заболевание «нейродегенерация с отложением железа в мозге 1» (англ. neurodegeneration with brain iron accumulation 1). В мире распространено сокращённое название заболевания как «NBIA1».
Примечания
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- Пономарев В.В. Болезнь Галлервордена — Шпатца (Клинический обзор и собственное наблюдение). Новости медицины и фармации. Международный неврологический журнал 3 (41) (2011). Дата обращения: 27 сентября 2017.
Литература
- Hochspringen ↑ Zhou u.a.: A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. in: Nature genetics (Nat Genet.). New York 28.2001,4 (Aug), 345—349. PMID 11479594 ISSN 1061-4036
- Hochspringen ↑ PKAN-Artikel bei genereviews von Gregory/Hayflick, revidierte Version Jan. 2008
- Hochspringen ↑ M. Shevell: Hallervorden and history. in: The New England journal of medicine (N Engl J Med.). Waltham Mass 2.2003,1 (Jan),348,3-4. PMID 12510036 ISSN 0028-4793