AutoDock
AutoDock — пакет программ, предназначенный для автоматизированного молекулярного докинга. В основном применяется для белок-лигандного докинга, в том числе, с учётом подвижных остатков белка. Autodock также используется для «слепого докинга», когда активный центр белка не известен.
| AutoDock | |
|---|---|
 | |
| Тип | Молекулярное моделирование |
| Разработчик | Scripps Research Institute |
| Написана на | C / C++ |
| Операционная система | Windows, macOS, Linux, Solaris |
| Последняя версия | 4.2.6 (2012-08-04) |
| Состояние | активное |
| Лицензия | GPL2+ (AutoDock 4) / ASL 2.0 (AutoDock Vina) |
| Сайт | autodock.scripps.edu |
О программе
AutoDock — один из пакетов программ, позволяющих предсказывать связывание маленьких молекул с белками известной структуры. Настоящие дистрибутивы AutoDock включают в себя два поколения ПО: AutoDock 4 и AutoDock Vina. AutoDock является свободным программным обеспечением, последняя 4-я версия которого распространяется в соответствии с открытым лицензионным соглашением GNU General Public License, AutoDock Vina доступна под Apache license[1][2].
Начиная с самой первой версии, Autodock представляет собой совокупность 2х программ: Autodock — собственно программа, проводящая докинг и Autogrid — программа, позволяющая рассчитывать сетки потенциалов. Для каждого рецептора (рецептором в докинге называется макромолекула, для которой рассчитываются сетки) достаточно рассчитать сетки потенциалов для каждого типа атомов один раз, что позволяет запускать расчеты для любого лиганда, состоящего из этих атомов (лиганд в докинге это маленькая молекула, для которой возможно изменение положения и конформации)[1].
В 2006 г. AutoDock был наиболее цитируемой программой для докинга[3].
AutoDock поддерживается и разрабатывается лабораториями The Scripps Research Institute и Olson Laboratory[1].
Механизм работы
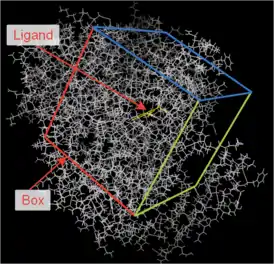
Докинг проводится в кубическую область внутри рецептора (docking box). С помощью AutoGrid для рецептора создается набор бинарных файлов — сеток потенциалов. Они описывают для каждого атома, входящего в docking box, потенциал его взаимодействия с пробным атомом определённого химического элемента. Набор этих элементов определяется химическим составом лигандов, с которыми требуется провести докинг. На каждый химический элемент создается по 1-2 файла. Для расчета потенциала используется скоринг функция, либо основанная на физических законах, либо эмпирическая, либо смешанная. Скоринг функции в разных версиях программы могут отличаться. Сетки потенциалов используются для расчета свободной энергии[4].
Лиганд, помимо совокупности атомов, связей и зарядов, внутри программы описывается набором чисел — положением в docking box, поворотом всех активных торсионных углов. Autodock перебирает все возможные комбинации этих чисел, чтобы в итоге найти оптимальное с точки зрения свободной энергии положение лиганда в docking box. Поэтому в качестве docking box обычно выбирают куб со стороной 10-30 ангстрем таким образом, чтобы он включал в себя активный центр рецептора[4].
Полный перебор всех возможных положений в активном центре и всех конформаций лиганда — очень время- и ресурсо-затратная задача. Для оптимизации процесса в Autodock применяются алгоритмы поиска глобального минимума: Монте-Карло, имитации отжига, генетические алгоритмы: LGA (Ламаркианский генетический алгоритм)[4].
Пример работы Autodock 4.2 на основе LGA[5]:
- Генерируется популяция лигандов (размер популяции задается пользователем). Каждый лиганд случайным образом расположен в docking boх, имеет случайную конформацию и описывается набором трансляционных, ориентационных и конформационных чисел.
- Для каждого элемента популяции рассчитывается свободная энергия. Она включает в себя энергию межмолекулярного взаимодействия рецептора с лигандом, и внутримолекулярную энергию лиганда.
- Стадия размножения. Для каждого элемента рассчитывается количество потомков по формуле: , где – количество потомков, – свободная энергия элемента, – энергия худшего элемента (максимальная), – средняя энергия. После этого каждый элемент лучше среднего получает пропорциональное количество потомков, которые добавляются в популяцию.
- Стадия кроссинговера: случайным образом выбираются два элемента популяции и их характеристические числа рекомбинируют, то есть элементы обмениваются некоторыми из чисел. Процент рекомбинирующих пар задается пользователем.
- Стадия мутации: к характеристическим числам элемента прибавляется случайное число, соответствующее распределению Коши. Параметры распределения и процент мутаций задаются пользователем.
- Стадия вымирания: худшие элементы популяции удаляются. Количество выживших также задается пользователем.
- Стадия оптимизации. Каждый элемент популяции оптимизируется с помощью алгоритмов локального поиска, например, имитации отжига при низких температурах, что позволяет минимизировать свободную энергию в небольших пределах. Эта стадия отличает LGA от классических генетических алгоритмов.
- Шаги 2-7 повторяются определённое большое количество раз (пока лучшая свободная энергия не перестанет изменяться, либо при достижении лимита по времени или количеству итераций).
- Вся предыдущая последовательность шагов выполняется несколько раз (независимые запуски).
- Результаты множественных запусков кластеризуются по положению в docking box с отсечкой по RMSD 1 ангстрем.
- Для всех находок в кластере считается энергия взаимодействия с рецептором. Энергией кластера считается самая лучшая в этом кластере.
- Финальная энергия рассчитывается для лучших кластеров как разность между разностями энергий перехода рецептора с лигандом из несвязанного положения в связанное[6].





История версий
Autodock 1
Создан в 1990 году. Первая версия базируется на силовом поле Amber. Скоринг функция является суммой потенциала Леннарда-Джонса, электростатического потенциала и эмпирических функций энергии ковалентных связей, плоских и торсионных углов[7].

В реализации доступны алгоритмы Монте-Карло и имитации отжига[8].
Autodock 2.4
Появилось много улучшений, сопутствующих программ для параллельных запусков. Новый алгоритм для улучшения поиска, в случае лигандов с большим количеством степеней свободы, основан на принципе разделяй и властвуй. Улучшен алгоритм локальной оптимизации[9].
Autodock 3
Для оптимизации поиска впервые был применен комбинированный генетический алгоритм LGA[10]. Новая полуэмпирическая скоринг функция, калибрующаяся на 30 белок-лигандных комплексах, учитывает направленные водородные связи и энергию сольватации[5].

Autodock 4, 4.2
Добавлен учёт возможной подвижности боковых цепей. Это достигнуто за счет разделения белка в два файла. Одна часть считается статичной, другая подвижной. Со статической частью работают с помощью подсчета энергии AutoGrid, с подвижной работают теми же методами, что и с лигандом. Созданы новые типы атомов, например, галогены и основные ионы металлов. Улучшена скоринг функция. Калибровка на 250 структурах из PDBBind. Появился AutoDockTools — специальное программное обеспечение, для подготовки файлов к докингу[6].
Autodock 4.2.5
Добавлена более прозрачная опция контроля вывода программы, позволяющая делать небольшой вывод для скрининга, и выводить отчет для более глубокого анализа. Некоторые ошибки при исполнении программы, выводившие предупреждение в предыдущих версиях, теперь останавливают программу. Это сделано в ответ на нужды скрининга, при котором пользователь может не заметить появляющихся предупреждений. Учёт электростатических взаимодействий между не связанными атомами в лиганде теперь по умолчанию включен. Также включение/отключение возможно при помощи команды intelec on/off[4].
Autodock Vina
AutoDock Vina является новым поколением ПО, разработанным Molecular Graphics Lab. Показывает значительные улучшения средних показателей accuracy-метрики в предсказании сайтов связывания, так же заметно увеличение скорости работы в два раза, по сравнению с AutoDock 4.1. Принципиально новая скоринг функция, основанная на алгоритме X-Score[11], которая разработана с учётом развития мультипроцессорных систем. За счет различий в погрешностях и самих используемых функций скоринга, используемых в AutoDock 4 и AutoDock Vina, программы могут показывать различные результаты на одних данных[12].
AutoDockTools
Существует ассоциированный с Autodock графический интерфейс AutoDockTools (ADT), который помогает анализировать докинг и выбирать связи в лиганде, которые будут считаться подвижными. Ниже упомянуты некоторые из функций ADT[1]:
- Видеть и вращать молекулы в формате 3D;
- Добавлять молекулы водорода, как полярные, так и не полярные;
- Ассоциировать частичные заряды атомов с лигандами и макромолекулами;
- Обозначать подвижные связи в лиганде, с использованием графической версии AutoTors;
- Настраивать DPF, GPF файлы;
- Запускать AutoGrid и AutoDock;
- Выводить и визуализировать результат работы AutoDock.
Входные файлы
Файлы с расширением .pdb являются основным форматом хранения информации о конформации и строении молекул. Такие файлы получают из экспериментальных данных полученных рентгеновской кристаллографией или ЯМР-спектроскопией, либо методами предсказания структуры молекул. Начиная с версии AutoDock 4 процедура докинга требует два файла в формате .pdbqt; один для рецептора, другой для лиганда. Если в белке необходимо учитывать подвижность некоторых аминокислот, то создается третий файл, содержащий информацию об атомах в подвижных частях белка. Преобразование файлов .pdb в формат .pdbqt возможно с помощью программы AutoDock Tools[4].
Файлы pdbqt содержат следующую информацию[4]:
- Координаты атомов,
- Полярные водороды,
- Частичные заряды на атомах,
- Тип атомов, например, есть два разных типа для алифатического и ароматического углерода,
- Сведения о подвижных связях.
Интерпретация результатов
В результате работы Autodock для одного лиганда получается dlg файл. В нём записан подробный отчет о работе программы. Он содержит в себе результаты каждого конкретного запуска с конечным положением лиганда (структура лиганда записана в формате pdbqt), рассчитанной энергией, затраченным на расчет временем[4].
Также доступен результат кластеризации: для каждого кластера показана населенность (Num in cluster), лучшая энергия (Lowest binding energy), к какому конкретно запуску она относится (Run)[4]:
CLUSTERING HISTOGRAM
____________________
________________________________________________________________________________
| | | | |
Clus | Lowest | Run | Mean | Num | Histogram
-ter | Binding | | Binding | in |
Rank | Energy | | Energy | Clus| 5 10 15 20 25 30 35
_____|___________|_____|___________|_____|____:____|____:____|____:____|____:___
1 | -3.44 | 150 | -3.44 | 2 |##
2 | -3.42 | 63 | -3.41 | 42 |#x42
3 | -3.42 | 187 | -3.40 | 83 |#x83
4 | -3.38 | 115 | -3.36 | 33 |#x33
5 | -3.32 | 128 | -3.31 | 37 |#x37
6 | -3.28 | 122 | -3.27 | 3 |###
_____|___________|_____|___________|_____|______________________________________Собственно результатами являются энергия ΔG и положение лиганда в активном центре рецептора. Разница энергий лигандов в первом приближении показывает, насколько один лиганд лучше связывается рецептором, чем другой. Положение лиганда в активном центре позволяет предсказывать механизм связывания[13].
Области применения
AutoDock находит применение в следующих областях[1]:
- Дизайн лекарств,
- Исследования химических процессов.
Autodock широко применяется в научном сообществе как для молекулярного докинга, так и для виртуального скрининга больших библиотек соединений (например ZINC)[14].
Докинг используется также для поиска блокаторов ферментов патогенных организмов, в частности топоизомеразы I туберкулезной палочки[15].
Белок тирозинфосфатазa В Mycobacterium tuberculosis (Mtb) (MptpB) является важным фактором вирулентности для бактерии, который способствует выживанию бактерий в макрофагах. Отсутствие человеческого ортолога делает MptpB привлекательной мишенью для новых терапевтических средств для лечения туберкулеза. Ингибиторы MptpB могут быть эффективным средством для преодоления возникающей лекарственной устойчивости к туберкулезу. Использовав стратегию виртуального скрининга на основе структуры, авторы успешно идентифицировали лекарственный ингибитор MptpB на основе тиобарбитурата[16].
С помощью Autodock нашли ингибиторы ВИЧ протеазы[17]. В частности, ингибиторы аспарагиновой пептидазы ВИЧ (HIV IPs) являются хорошими кандидатами для повторного использования лекарственных средств.
Кроме того, докинг используется для поиска лигандов, взаимодействующих с транскрипционными факторами. Например, HNF-1a является транскрипционным фактором, который регулирует метаболизм глюкозы путем экспрессии в различных тканях. С помощью докинга in silico были найдены потенциальные мишени[18].
С помощью докинга были смоделированы взаимодействия двух регуляторов транскрипции, ExuR и UxuR, с субстратами и интермедиатами гликолиза, путей Эшвелла и Энтнера-Дудорова. Для UxuR были обнаружены два предпочтительных сайта связывания лиганда: один расположен в С-терминальном домене, а другой занимает междоменное пространство. Для ExuR в междоменной области был обнаружен только один предпочтительный сайт[19].
Autodock (в основном Vina) широко применяется в большом числе автоматических систем виртуального скрининга[20][21][22].
Сотрудничество с проектами
World Community Grid предлагает свою помощь в получении бесплатной вычислительной мощности для ускорения исследований, проводимых на основе AutoDock[1].
AutoDock запущен в работу на основе World Community Grid со следующими проектами:
- FightAIDS@Home от The Scripps Research Institute[23],
- Discover Dengue Drugs — Together от The University of Texas Medical Branch[24],
- Help Fight Childhood Cancer[25],
- Influenza Antiviral Drug Search[26],
- GO Fight Against Malaria[27].
Альтернативные программы
В 2016 году была проведена оценка различных программ на выборке из 2002 комплексов белка с лигандом. Оценивалась частота совпадений в найденных с помощью докинга позициях. Совпадением считались случаи, когда RMSD между найденным и нативным положением лиганда не превышало 2 Å[28][29].
Среди альтернативных академических программ выделяют LeDock, rDock, UCSF Dock, при этом первая программа показала наилучший результат (57,4% совпадения)[29].
Коммерческие альтернативные программы показали лучший результат (59,8% совпадения у GOLD), чем академические. Также в исследовании участвовали программы Surflex, FlexX, Glide, LigandFit, MOE-Dock, ICM_pro, MCDock, FRED.[28].
| Программа | Совпадение | Программа | Совпадение | |
|---|---|---|---|---|
| GOLD | 59,8 % | Autodock Vina | 49,0 % | |
| Glide (XP) | 57,8 % | AutoDock (PSO) | 47,3 % | |
| LeDock | 57,4 % | LigandFit | 46,1 % | |
| Glide (SP) | 53,8 % | MOE Dock | 45,6 % | |
| Surflex-Dock | 53,2 % | UCSD DOCK | 44,0 % | |
| rDock | 50,3% | AutoDock (LGA) | 37,4 % |
Примечания
- Garrett M. Morris. AutoDock (англ.). autodock.scripps.edu. Дата обращения: 4 апреля 2017.
- AutoDock Vina - molecular docking and virtual screening program. http://vina.scripps.edu.+Дата обращения: 3 мая 2019.
- Sousa Sérgio Filipe, Fernandes Pedro Alexandrino, Ramos Maria João. Protein-ligand docking: Current status and future challenges (англ.) // Proteins: Structure, Function, and Bioinformatics. — 2006. — 21 July (vol. 65, no. 1). — P. 15—26. — ISSN 0887-3585. — doi:10.1002/prot.21082.
- Garrett M. Morris, David S. Goodsell, Michael E. Pique, William “Lindy” Lindstrom, Ruth Huey, Stefano Forli, William E. Hart, Scott Halliday, Rik Belew and Arthur J. Olson. AutoDock4.2 User Guide.
- doi:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
Вы можете подставить цитату вручную или с помощью бота. - Morris Garrett M., Huey Ruth, Lindstrom William, Sanner Michel F., Belew Richard K., Goodsell David S., Olson Arthur J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility (англ.) // Journal of Computational Chemistry. — 2009. — December (vol. 30, no. 16). — P. 2785—2791. — ISSN 0192-8651. — doi:10.1002/jcc.21256.
- Goodford P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. (англ.) // Journal Of Medicinal Chemistry. — 1985. — July (vol. 28, no. 7). — P. 849—857. — PMID 3892003.
- Goodsell David S., Olson Arthur J. Automated docking of substrates to proteins by simulated annealing (англ.) // Proteins: Structure, Function, and Genetics. — 1990. — Vol. 8, no. 3. — P. 195—202. — ISSN 0887-3585. — doi:10.1002/prot.340080302.
- Morris Garrett M., Goodsell David S., Huey Ruth, Olson Arthur J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4 (англ.) // Journal of Computer-Aided Molecular Design. — 1996. — August (vol. 10, no. 4). — P. 293—304. — ISSN 0920-654X. — doi:10.1007/BF00124499.
- Fuhrmann Jan, Rurainski Alexander, Lenhof Hans-Peter, Neumann Dirk. A new Lamarckian genetic algorithm for flexible ligand-receptor docking (англ.) // Journal of Computational Chemistry. — 2010. — P. NA—NA. — ISSN 0192-8651. — doi:10.1002/jcc.21478.
- Wang R., Lai L., Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. (англ.) // Journal Of Computer-aided Molecular Design. — 2002. — January (vol. 16, no. 1). — P. 11—26. — PMID 12197663.
- Trott Oleg, Olson Arthur J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading (англ.) // Journal of Computational Chemistry. — 2009. — P. NA—NA. — ISSN 0192-8651. — doi:10.1002/jcc.21334.
- Ma Xiaoli, Yan Jin, Xu Kailin, Guo Luiqi, Li Hui. Binding mechanism of trans-N-caffeoyltyramine and human serum albumin: Investigation by multi-spectroscopy and docking simulation (англ.) // Bioorganic Chemistry. — 2016. — June (vol. 66). — P. 102—110. — ISSN 0045-2068. — doi:10.1016/j.bioorg.2016.04.002.
- Anand R. Identification of Potential Antituberculosis Drugs Through Docking and Virtual Screening. (англ.) // Interdisciplinary Sciences, Computational Life Sciences. — 2018. — June (vol. 10, no. 2). — P. 419—429. — doi:10.1007/s12539-016-0175-6. — PMID 27147082.
- Sandhaus Shayna, Chapagain Prem P., Tse-Dinh Yuk-Ching. Discovery of novel bacterial topoisomerase I inhibitors by use of in silico docking and in vitro assays (англ.) // Scientific Reports. — 2018. — 23 January (vol. 8, no. 1). — ISSN 2045-2322. — doi:10.1038/s41598-018-19944-4.
- Zhang D., Lin Y., Chen X., Zhao W., Chen D., Gao M., Wang Q., Wang B., Huang H., Lu Y., Lu Y. Docking- and pharmacophore-based virtual screening for the identification of novel Mycobacterium tuberculosis protein tyrosine phosphatase B (MptpB) inhibitor with a thiobarbiturate scaffold. (англ.) // Bioorganic Chemistry. — 2019. — April (vol. 85). — P. 229—239. — doi:10.1016/j.bioorg.2018.12.038. — PMID 30641319.
- Schames Julie R., Henchman Richard H., Siegel Jay S., Sotriffer Christoph A., Ni Haihong, McCammon J. Andrew. Discovery of a Novel Binding Trench in HIV Integrase (англ.) // Journal of Medicinal Chemistry. — 2004. — April (vol. 47, no. 8). — P. 1879—1881. — ISSN 0022-2623. — doi:10.1021/jm0341913.
- Sridhar Gumpeny Ramachandra, Nageswara Rao Padmanabhuni Venkata, Kaladhar Dowluru SVGK, Devi Tatavarthi Uma, Kumar Sali Veeresh. In SilicoDocking of HNF-1a Receptor Ligands (англ.) // Advances in Bioinformatics. — 2012. — Vol. 2012. — P. 1—5. — ISSN 1687-8027. — doi:10.1155/2012/705435.
- Tutukina Maria N., Potapova Anna V., Vlasov Peter K., Purtov Yuri A., Ozoline Olga N. Structural modeling of the ExuR and UxuR transcription factors ofE. coli: search for the ligands affecting their regulatory properties (англ.) // Journal of Biomolecular Structure and Dynamics. — 2016. — 6 January (vol. 34, no. 10). — P. 2296—2304. — ISSN 0739-1102. — doi:10.1080/07391102.2015.1115779.
- Wójcikowski Maciej, Zielenkiewicz Piotr, Siedlecki Pawel. Open Drug Discovery Toolkit (ODDT): a new open-source player in the drug discovery field (англ.) // Journal of Cheminformatics. — 2015. — 22 June (vol. 7, no. 1). — ISSN 1758-2946. — doi:10.1186/s13321-015-0078-2.
- AutoDock | Raccoon2. autodock.scripps.edu. Дата обращения: 15 мая 2016.
- Sharma V., Pattanaik K. K., Jayprakash V., Basu A., Mishra N. A utility script for automating and integrating AutoDock and other associated programs for virtual screening. (англ.) // Bioinformation. — 2009. — 6 September (vol. 4, no. 2). — P. 84—86. — PMID 20198176.
- FightAIDS@Home. www.worldcommunitygrid.org. Дата обращения: 31 марта 2017.
- Discovering Dengue Drugs – Together. www.worldcommunitygrid.org. Дата обращения: 31 марта 2017.
- Help Fight Childhood Cancer. www.worldcommunitygrid.org. Дата обращения: 31 марта 2017.
- Influenza Antiviral Drug Search. www.worldcommunitygrid.org. Дата обращения: 31 марта 2017.
- Rarey. A fast flexible docking method using an incremental construction algorithm.
- Wang Zhe, Sun Huiyong, Yao Xiaojun, Li Dan, Xu Lei, Li Youyong, Tian Sheng, Hou Tingjun. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power (англ.) // Physical Chemistry Chemical Physics. — 2016. — Vol. 18, no. 18. — P. 12964—12975. — ISSN 1463-9076. — doi:10.1039/c6cp01555g.
- Pagadala Nataraj S., Syed Khajamohiddin, Tuszynski Jack. Software for molecular docking: a review (англ.) // Biophysical Reviews. — 2017. — 16 January (vol. 9, no. 2). — P. 91—102. — ISSN 1867-2450. — doi:10.1007/s12551-016-0247-1.