Фибродисплазия
Фибродисплазия оссифицирующая прогрессирующая или ФОП (от лат. fibrō «волокно» + dis «расстройство, нарушение» + др.-греч. πλάσις «строение, структура»; лат. os, ossis «кость» + facio «делать» = «окостенение») — «мягкая соединительная ткань, которая прогрессивно превращается в кость». Очень редкое и тяжёлое по своему течению генетическое заболевание, при котором мышцы, сухожилия и связки постепенно превращаются в кости. Процесс прогрессирует с годами, начинаясь обычно в пределах десятилетнего возраста у детей с мутацией определенного гена. Самые ранние зарегистрированные случаи относятся ко времени XVII и XVIII столетий. В 1692 французский врач Гай Пэтин встретился с пациентом, который имел ФОП и упоминал встречу в своих письмах. В 1736, британец врач Джон Фрек описал подробно подростка, диагноз которого включал набухания по всей спине.
| Фибродисплазия оссифицирующая прогрессирующая | |
|---|---|
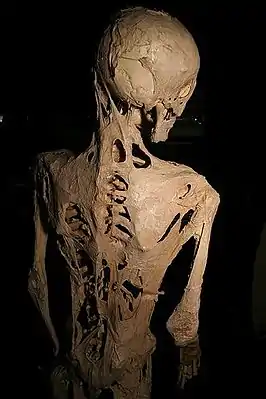 Скелет человека, страдавшего ФОП | |
| МКБ-11 | FB31.1 |
| МКБ-10 | M61.1 |
| МКБ-10-КМ | M61.10 и M61.1 |
| МКБ-9 | 728.11 |
| МКБ-9-КМ | 728.11[1] |
| OMIM | 135100 |
| DiseasesDB | 8732 |
| eMedicine | derm/609 |
| MeSH | D009221 |
Болезнь стала известной как миозит оссифицирующий прогрессирующий, что означает «превращение мышцы прогрессивно в кость». Название было официально изменено на фибродисплазию оссифицирующую прогрессирующую в 1970-х доктором Виктором Маккьюзиком из Школы медицины Университета имени Джонса Хопкинса, которого считают отцом современной медицинской генетики, в качестве обоснования он привел то что другие мягкие (или волокнистые — fibro) ткани в дополнение к мышцам (например, сухожилия и связки) могут быть затронуты окостенением.
Также заболевание относится к врожденной наследственной патологии с аутосомно - доминантным типом наследования. Оно характеризуется неуклонно прогрессирующим течением, приводит к значительным нарушениям функционального состояния опорно-двигательного аппарата, глубокой инвалидизации больных и преждевременной их смерти, причем преимущественно в детском и молодом возрасте. Основу фибродисплазии составляет формирование воспалительных процессов в сухожилиях, связках, фасциях, апоневрозах и мышцах, что в конечном итоге приводит к их кальцификации и окостенению. Болезнь ещё называют «Болезнь второго скелета», так как там, где в организме должны происходить штатные противовоспалительные процессы, начинается рост кости. У болезни нет расовой, половой, географической предрасположенности. Чаще всего болезнь возникает как результат спонтанной новой мутации.
Симптомы
Дети, рожденные с фибродисплазией, отличаются характерной патологией большого пальца ноги - одна или несколько фаланг пальца искривлены вовнутрь и иногда в нем не хватает сустава. Этот палец и дает 95-процентную вероятность диагностирования заболевания у ребенка. Также болезнь характеризуется обострениями, как правило, без видимых причин. Обострения бывают нескольких видов, самое распространенное — появление под кожей ребенка уплотнений размером от одного до десяти сантиметров неопределимого характера, на шее, спине, предплечьях в возрасте до 10 лет, в любом другом месте в более старшем возрасте. Одним из признаков является отек мягких тканей головы, как при незначительном повреждении (ушиб, укус насекомого, царапина), так и при сильных травмах. Отек не спадает до месяца, не реагирует ни на какую медикаментозную терапию. На месте уплотнений могут возникать оссификации, которые не поддается ни в каком лечении и не поддаются никаким препаратам. Болезнь часто путают с раком и другими заболеваниями, подобные затвердения пытаются удалять, что приводит к ещё более бурному росту «лишних» костей, и является основной причиной инвалидизации страдающих этим заболеванием на данный момент.
Исследования на всех уровнях предполагают участие и реакцию иммунной системы в ФОП. Присутствие макрофагов, лимфоцитов и тучных клеток на ранних стадиях ФОП. Макрофаги и лимфоциты в пораженной болезнью мускулатуре, обострения после вирусной инфекции, спонтанность наступления обострений, и чувствительность ранних обострений к кортикостероидам, убеждают нас в причастности иммунной системы в патогенезе нарушений в ФОП. ФОП характеризуется лентовидными, пластиноподобными, прослоечными кальцификациями и заменяет скелетные мускулы и соединительные ткани через процесс внутрихрящевого окостенения, что приводит к опоясывающему конкретный регион окостенению и постоянной неподвижности. ФОП типично начинается в шейном отделе, мышцах спины, около позвоночника, на голове, в плечевых суставах и соседствующих областях, позже болезнь опускается в брюшной, бедренный отдел и отдалённые от центра области. Несколько скелетных мускулов включая диафрагму, язык, и около-глазные мускулы не могут быть затронуты ФОП. Сердечная мускулатура и гладкая мускулатура так же никогда не затрагивается болезнью. Формирование кости в ФОП является эпизодическим, но последствия этого всеобъемлющи.
Лечение
Вспышки ФОП являются спорадическими и непредсказуемыми, и есть огромная разница в каждом индивидуальном проявлении. Несколько базовых научных исследований подтвердили невозможность предсказать начало, силу, последствия и продолжительность обострения ФОП, хотя особенности анатомических патологий уже были описаны. Редкость ФОП и её непредсказуемая природа делает чрезвычайно трудным оценку эффективности любого терапевтического вмешательства, факт, признанный 1918 году врачом Джулиус Розенстирн :
«Болезнь развивается со всеми видами средств и альтернатив дефектного метаболизма; каждое отдельное терапевтическое вмешательство с более или менее отмеченным успехом, наблюдаемым исключительно его оригинальным автором, в будущем раскритиковывается любым последователем. Во многих случаях, признаки болезни исчезают часто спонтанно, таким образом, терапевтический эффект (любого лечения) не может быть полностью подтвержден своей эффективностью.»
Джулиус Розенстирн
В настоящее время не существует никаких доказанных эффективных методов профилактики или лечения ФОП. С открытием гена ФОП стало возможным понимание патогенеза заболевания. Перспективной является непосредственная работа с геномом, но в настоящее время данные методы лечения находятся лишь на стадии предварительных лабораторных исследований и не применяются в клинической практике.
Генетический аспект и будущее лечение
В 2006 году исследовательская группа из Университета штата Пенсильвания открыла ген, мутация в котором приводит к этому заболеванию. Чтобы идентифицировать хромосомное местоположение для гена ФОП, консервативной связки всего генома, анализ проводился с помощью наблюдения за пятью семьями с самыми однозначными особенностями ФОП. Этот подход идентифицировал связку ФОП к хромосоме 2q23-24.
Генотерапия: В настоящее время идет работа над генными блокаторами мутации в гене ACVR1/ALK2.В декабре 2012 продолжаются опыты по лечению гетеротопической оссификации лабораторным мышам. Препарат показал свою действенность как в генетически обусловленной так и в посттравматической оссификации. Золотой стандарт для всех исследований нового препарата — тройной слепой перекрестный метод с применением плацебо, но такие исследования будет достаточно трудно провести при ФОП, ввиду малого количества пациентов с болезнью в уже развитой стадии, неоднозначным патогенезом болезни, а также большой разницей в проявлениях обострений у пациентов. Ввиду того, что фибродисплазия является чрезвычайно редким заболеванием, с разной тяжестью проявлений, экспериментальным методам лечения предстоит пройти серьёзную оценку на предмет дозировки и продолжительности курса.
14 июля 2014 года начата фаза 2 клинических исследований на людях препарата "Паловаротен"[2]. Исследования проводятся в Сан-Франциско, Филадельфии и Париже.
Примечания
- база данных Disease ontology (англ.) — 2016.
- An Efficacy and Safety Study of Palovarotene to Treat Preosseous Flare-ups in FOP Subjects - Full Text View - ClinicalTrials.gov (англ.). clinicaltrials.gov. Дата обращения: 6 мая 2019.
Ссылки
- Сайт российской межрегиональной организации помощи пациентам с заболеванием фибродисплазия "Живущие с ФОП и их друзья"
- The International FOP Association Международная ассоциация ФОП
- Михалева Г. В., Сермягина И. Г., Геппе Н. А., Рябова Т. В. Клинические и рентгенологические проявления оссифицирующей прогрессирующей фибродисплазии у детей.