Сборка генома
Сборка генома — процесс объединения большого количества коротких фрагментов ДНК (ридов) в одну или несколько длинных последовательностей (контигов и скаффолдов) в целях восстановления последовательностей ДНК хромосом, из которых возникли эти фрагменты в процессе секвенирования.
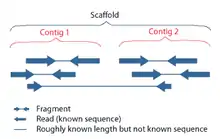
Сборка генома является очень сложной вычислительной задачей, в частности, осложнённой тем, что геномы часто содержат большое количество одинаковых повторяющихся последовательностей (так называемые геномные повторы). Эти повторы могут быть длиной в несколько тысяч нуклеотидов, а также встречаться в тысяче различных мест в геноме. Особенно богаты повторами большие геномы растений и животных, в том числе геном человека.
Алгоритмические подходы
Существует два подхода для сборки геномов — основанный на перекрытии overlap-layout-consensus (применяется для длинных фрагментов), а также основанный на графах де Брёйна (применяется для коротких фрагментов) [1][2].
Overlap-Layout-Consensus
При секвенировании методом дробовика все ДНК организма сначала разрезают на миллионы маленьких фрагментов до 1000 нуклеотидов в длину. Затем алгоритмы сборки генома рассматривают полученные фрагменты одновременно, находя их перекрытия (overlap), объединяя их по перекрытиям (layout) и исправляя ошибки в объединённой строке (consensus). Данные шаги могут повторяться несколько раз в процессе сборки.
Данный подход был наиболее распространён для сборки геномов до появления секвенирования следующего поколения.
Графы де Брёйна
С развитием технологий секвенирования следующего поколения получение фрагментов стало на порядок дешевле, но размер фрагментов стал меньше (до 150 нуклеотидов), а количество ошибок при чтении фрагментов увеличилось (до 3 %). При сборке таких данных получили распространение методы[3], основанные на графах де Брёйна.
Доступные сборщики
Список популярных геномных сборщиков:
| Название | Поддерживаемые технологии | Авторы | Представлен | Обновлён | Лицензия* | Домашняя страница |
|---|---|---|---|---|---|---|
| ABySS | Solexa, SOLiD | Simpson, J. et al. | 2008 | 2011 | NC-A | ссылка |
| ALLPATHS-LG | Solexa, SOLiD | Gnerre, S. et al. | 2011 | 2011 | OS | ссылка |
| CLC Genomics Workbench | Sanger, 454, Solexa, SOLiD | CLC bio | 2008 | 2010 | C | ссылка |
| Euler | Sanger, 454 (,Solexa ?) | Pevzner, P. et al. | 2001 | 2006 | (C / NC-A?) | ссылка |
| Euler-sr | 454, Solexa | Chaisson, MJ. et al. | 2008 | 2008 | NC-A | ссылка |
| IDBA | Sanger,454,Solexa | Yu Peng, Henry C. M. Leung, Siu-Ming Yiu, Francis Y. L. Chin | 2010 | 2010 | (C / NC-A?) | ссылка |
| MIRA | Sanger, 454, Solexa | Chevreux, B. | 1998 | 2011 | OS | ссылка |
| Newbler | 454, Sanger | 454/Roche | 2009 | 2009 | C | ссылка |
| SOPRA | Illumina, SOLiD, Sanger, 454 | Dayarian, A. et al. | 2010 | 2011 | OS | ссылка |
| SOAPdenovo | Solexa | Li, R. et al. | 2009 | 2009 | OS | ссылка |
| SPAdes | Illumina, Solexa | Bankevich, A et al. | 2012 | 2012 | OS | ссылка |
| Velvet | Sanger, 454, Solexa, SOLiD | Zerbino, D. et al. | 2007 | 2009 | OS | ссылка |
| Canu | PacBio, Oxford Nanopore | Koren, S. et al. | 2017 | 2020 | OS | ссылка |
| *Licences: OS = Open Source; C = Коммерческая; C / NC-A = Коммерческая, но бесплатна для использования в некоммерческих и научных целях; Скобки = неизвестно, но скорее всего C / NC-A | ||||||
Примечания
- Zhenyu Li et al. Comparison of the two major classes of assembly algorithms: overlap–layout–consensus and de-bruijn-graph (англ.) // Briefings in Functional Genomics : journal. — 2012. — Vol. 11, no. 1. — P. 25—37. — doi:10.1093/bfgp/elr035.
- Miller J. R., Koren S., Sutton G. Assembly algorithms for next-generation sequencing data (англ.) // Genomics : journal. — Academic Press, 2010. — Vol. 95, no. 6. — P. 315—327.
- Pavel A. Pevzner, Haixu Tang, Michael S. Waterman. An Eulerian path approach to DNA fragment assembly (англ.) // Proceedings of the National Academy of Sciences of the United States of America : journal. — 2001. — Vol. 98, no. 17. — P. 9748—9753. — doi:10.1073/pnas.171285098.