Биоаналог
Биоаналог (англ. biosimilar; также называемый аналогичный биологический лекарственный препарат, биоаналогичный лекарственный препарат, биосимиляр или биоподобный лекарственный препарат) — биологический препарат, являющийся почти идентичной копией (версией) оригинального препарата и производимый другой компанией.[1]. Биоаналоги — это официально одобренные версии оригинальных «инновационных» препаратов, их производство и выпуск на рынок возможны по истечении действия патента оригинального препарата.[2]. Подтверждение эквивалентности биоаналога в рамках комплекса сравнительных исследований с оригинальным препаратом по показателям физико-химическим характеристикам, биологической активности, Фармакодинамике и фармакокинетике, иммуногенности, а также клинической безопасности и эффективности является обязательным для получения разрешения на вывод на рынок; биоаналог должен создаваться на основе профиля физико-химических и биологических свойств оригинальной молекулы.
В отличие от воспроизведенных лекарств, содержащих низкомолекулярные соединения, биопрепаратам обычно характерна высокая сложность молекулярного строения и высокая чувствительность к модификациям и изменениям процессов их производства. Несмотря на такую гетерогенность молекулярного строения действующего вещества, все биопрепараты, включая биоаналоги, должны обладать такими качеством и клиническими характеристиками, которые были бы стабильны на протяжении всего их жизненного цикла.[3] Производители биоаналогов не имеют доступа к молекулярному клону оригинального разработчика и оригинальному банку клеток, к точным данным о процессе ферментации и очистки или к действующему лекарственному веществу, однако у них есть доступ к продаваемому инновационному продукту. По этой причине гораздо сложнее провести обратную инженерию (то есть создать всю цепочку производственных процессов, зная только характеристики конечного подукта) и достичь взаимозаменяемости между «воспроизведёнными» и инновационными биопрепаратами, нежели если бы они были лекарственными препаратами на основе полностью синтетических или полусинтетических лекарственных веществ. Именно поэтому было придумано слово «биоаналог» для разграничения между такими «воспроизведенными» биопрепаратами и низкомолекулярными генериками. Простая аналогия, часто используемая для объяснения разницы, состоит в сравнении вина и газировки. Объективно труднее признать «достаточную взаимозаменяемость» двух бутылок вина из двух винокурен в силу различий в штамме закваски, погодных условиях и годе сбора урожая, нежели признать «достаточную взаимозаменяемость» двух бутылок газировки с одинаковым ароматизатором, производимых на двух разных заводах, ибо они содержат одинаковый ароматизатор, свойства которого на обоих заводах идентичны.
Такие лекарственные ведомства, как Европейское агентство по лекарствам (EMA) Евросоюза, Администрация по продуктам питания и лекарствам (FDA) США и Департамент медицинских продуктов и продовольствия Здоровья Канады, разработали собственные указания, содержащие требования, предъявляемые к подтверждению аналогичности двух биологических препаратов с точки зрения безопасности и эффективности. В соответствии с этими указаниями, биологический препарат биоаналогичен референтному препарату, на основании данных, полученных по результатам (a) аналитических исследований, подтверждающих, что биологический препарат высоко аналогичен референтному препарату, невзирая не незначительные различия в клинически неактивных компонентах, (b) исследований на животных (включая оценку токсичности) и (c) клинического исследования или исследований (включая оценку иммуногенности и фармакокинетики или фармакодинамики). Комплекс данных должен быть достаточен для доказательства того, что отсутствуют клинически значимые различия между биологическим препаратом и референтным препаратом с точки зрения их безопасности, чистоты и активности при одном или более соответствующих показаниях, при которых разрешено применение референтного препарата, и для применения при которых планируется зарегистрировать биоаналогичный препарат.
Например, в случае такого лекарственного препарата, содержащего моноклональные антитела, как Ремзима, было выполнено обширное установление физико-химических и биологических характеристик разрабатываемого моноклонального антитела и его референтного препарата (Ремикейд), чтобы подтвердить высокую аналогичность их свойств. Высокая структурная аналогичность (вплоть до уровня совпадения по типу «отпечатков пальцев») позволяет надеяться, что функциональная активность воспроизводимого антитела будет такой же, как и у эталонного препарата. Вместе с тем, поскольку сегодняшний уровень знаний не позволяет полностью прогнозировать, какие различия в структурных характеристиках (а они всегда будут, поскольку даже оригинальный препарат от серии к серии незначительно отличается по своим структурным и функциональным характеристикам) будут приводить к клинически значимым различиям между двумя лекарственными препаратами, и проводится комплекс биологических испытаний, испытаний на животных и клинических исследований. Такие исследования в условиях in vitro («в пробирке») или in vivo (на живых животных или людях) направлены на оценку клинической значимости различий, выявленных по результатам обширных аналитических физико-химических испытаний. Важно при этом помнить, что клинические исследования биоаналогов не направлены на подтверждение эффективности и безопасности биоаналогичного лекарственного препарата при некотором заболевании. Основная цель таких исследований — подтвердить, что биоаналог столь же эффективен и безопасен, что и эталонный оригинальный биологический лекарственный препарат.[4]
По этой причине разработка и одобрение биоаналогов происходят гораздо медленнее. Так, если сравнивать с воспроизведёнными лекарственными препаратами, с 2006 г., когда вступило в силу новое законодательство о биоаналогичности, в Европейском союзе лишь небольшому числу биоаналогов было выдано разрешение на продажу. По состоянию на май 2019 г. в ЕС одобрено 54 биоаналога. В США по состоянию на май 2019 г. одобрено 19 биоаналогов.[5] При этом первое одобрение в США состоялось только 6 марта 2015 г., когда FDA выдала разрешение на биоаналогичный филграстим, получивший название филграстим-sndz (торговое название Zarxio), компании Сандоз.
Процессы одобрения
Регуляторные органы Евросоюза создали специальную процедуру одобрения для выдачи разрешений на последующие версии ранее одобренных биопрепаратов, назвав их «аналогичные биологические лекарственные препараты» или биоаналоги. Процедура основана на скрупулезном подтверждении «сопоставимости» «аналогичного» препарата по отношению к существующему одобренному препарату.[6] В Соединенных Штатах Администрация по продуктам питания и лекарствам (FDA) признала необходимость нового законодательства, чтобы получить возможность одобрять биоаналоги биопрепаратов, первоначально одобренных на основании Закона о службе здравоохранения.[7] Состоялись дополнительные слушания в Конгрессе.[8] 17 марта 2009 г. Палата представителей внесла инициативу о принятии Закона о биоаналогах.[2] См. веб-сайт Библиотеки Конгресса, поиск по «H.R. 1548» в 111-й сессии Конгресса. С 2004 г. FDA провела серию публичных заседаний, посвященных биоаналогам.[9][10]
FDA получила полномочия одобрять биоаналоги (включая взаимозаменяемые биопрепараты, которыми допустимо заменять соответствующий референтный препарат) в рамках Закона о защите пациентов и доступной помощи, подписанного президентом США Бараком Обамой 23 марта 2010 г.
FDA ранее одобряла биологические препараты на основании их сопоставимости, например, в мае 2006 г. был одобрен Omnitrope, однако подобно эноксапарину натрия, это был референтный препарат Genotropin, изначально одобренный в качестве биологического лекарства на основании Федерального закона о продуктах питания, лекарствах и косметике.[11]
6 марта 2015 г. Zarxio стал первым биоаналогом, одобренным FDA.[12] Zarxio компании Сандоз является биоаналогом препарата Нейпоген (филграстим) компании Амджен, который, свою очередь, был первоначально зарегистрирован в 1991 г. Он стал первым препаратом, получившим разрешение на основании Закона о ценовой конкуренции и инновациях биопрепаратов от 2009 г. (Закона о ЦКИБ), принятого в составе Закона о доступном здравоохранении. Однако, как отмечает FDA, Zarxio был одобрен в качестве биоаналога, а не взаимозаменяемого препарата. При этом согласно Закону о ЦКИБ лишь биопрепарат, одобренный в качестве «взаимозаменяемого», может заменять референтный препарат без вмешательства медицинского работника, назначившего референтный препарат. FDA указало, что одобрение агентством Zarxio основано на доказательствах, включавших установление структурных и функциональных характеристик, животных данных, фармакокинетических и фармакодинамических данных на человеке, данных клинической иммуногенности и других данных о клинической безопасности и эффективности, подтвердивших биоаналогичность Zarxio Нейпогену.
Справочные сведения
Клонирование генетического материала человека и разработка in vitro-биологических производственных систем открыли возможности для получения практически любого биологического вещества на основе рекомбинантной ДНК с целью последующей разработки лекарства. Технология моноклональных антител в комбинации с технологией рекомбинантной ДНК открыла дорогу для индивидуализированных и таргетных лекарств. Появляются генная и клеточная терапии в качестве еще более новых подходов.
Рекомбинантные терапевтические белки имеют сложное строение: состоят из длинной цепи аминокислот, модифицированных аминокислот, дериватизируемых углеводными остатками, свернутых с помощью сложных механизмов, приобретая тем самым комплексные структуры высоких порядков. Такие белки вырабатываются живыми клетками (бактериальными, дрожжевыми, животными, растительными или человеческими клеточными линиями либо клеточными линиями насекомых, а также трансгенными растениями или животными). Конечные характеристики лекарства, содержащего рекомбинантный терапевтический белок, во многом определяются процессом их получения: выбранным типом клеток, разработкой генетически модифицированной клетки для биосинтеза, созданием системы банков клеток из генетически модифицированного клона, наращиванием клеток из банка для получения клеток-продуцентов, процессом биосинтеза, осуществляемым клетками продуцентами, процессом очистки наработанного белка, формуляцией терапевтического белка в лекарство.
По истечении патента на одобренные рекомбинантные лекарства (например, инсулин, гормон роста человека, интерфероны, эритропоэтин, моноклональные антитела и т. д.) любая другая биотехнологическая компания вправе разработать и вывести на рынок эти биопрепараты (называемые по этой причине биоаналогами). Каждый биопрепарат проявляет некоторую степень вариабельности в своих структурных характеристиках и профиле примесей, даже между разными сериями одного и того же препарата, что объясняется неизбежной вариабельностью биологической экспрессирующей системы и производственным процессом.[13] Процесс производства любого референтного препарата подвергается многочисленным изменениям, причем такие изменения производственного процесса (простирающиеся от изменения поставщика питательной среды для клеточной культуры до новых методов очистки или организации производства на новых производственных площадках) подлежат одобрению на основании соответствующих данных и — при необходимости — инспектирования на месте регуляторными органами (например, FDA). В противоположность этому, в случае биоаналогов, помимо аналитических испытаний, обязательно проведение также доклинических и клинических исследований с использованием наиболее чувствительных экспериментальных моделей, позволяющих обнаружить различия между двумя препаратами с точки зрения клинической фармакокинетики (ФК) и фармакодинамики (PD), иммуногенности, безопасности и эффективности, которые были бы значимы с медицинской точки зрения. Эти исследования направлены на подтверждение, что невзирая на структурные и функциональные отличия от оригинального препарата, по клиническим характеристикам (по степени эффективности и профилю безопасности у пациентов) биоаналог достаточно схож с оригинальным препаратом. Вместе с тем клинические исследования биоаналогов не направлены на доказательство эффективности и безопасности биоаналогичного лекарственного препарата при некотором заболевании. Основная цель таких исследований — подтвердить, что биоаналог столь же эффективен и безопасен, что и эталонный оригинальный биологический лекарственный препарат.[4]
Текущая концепция разработки биоаналогичных моноклональных антител строится на принципе обширного физико-химического аналитического и функционального сравнения молекул, которое дополняется сравнительными доклиническими и клиническими данными, позволяющем установить эквивалентную эффективность и безопасность при «модельном» показании к применению, являющемся наибольшее чувствительным с точки зрения обнаружения любых незначительных различий (существуй они) между биоаналогичным и соответствующим референтным моноклональными антителами на клиническом уровне.
Европейское агентство по лекарствам (EMA) Евросоюза признает этот факт, который подвигнул агентство на создание понятия «биоаналог», чтобы подчеркнуть, что несмотря на то что биоаналогичные препараты аналогичны оригинальному препарату, они всё же не являются точной копией последнего.[14] Каждый биопрепарат проявляет некоторую степень вариабельности. Вместе с тем, если можно доказать сопоставимость биоаналога и референтного препарата по структуре и функциям, фармакокинетическим профилям и фармакодинамическим эффектам и (или) по эффективности, можно также ожидать, что нежелательные лекарственные реакции, обусловленные избыточными фармакологическими эффектами, будут иметь схожую частоту возникновения.
Первоначально сложность биологических молекул влекла за собой необходимость предоставления обширных данных о безопасности и эффективности биоаналога в целях его одобрения. Постепенно взамен них стали все больше полагаться на анализы (начиная с фармацевтических и заканчивая клиническими), обладающие аналитической чувствительностью, достаточной для обнаружения любых существенных различий в дозе.[15] Это объясняется все большими достижениями науки и техники, которые улучшают наше понимание зависимости между структурой макромолекулярных комплексов и их вариациями с одной стороны и функциями, определяемыми такими структурами, — с другой. Вместе с тем безопасное применение биопрепаратов зависит от информированного и правильного применения медицинскими работниками и пациентами, поскольку биомолекулы более чувствительны к внешним условиям, например, к свету, не выдерживают встряхивания, избирательны по отношению к системам для введения в организм. Внедрение биоаналогов также требует специально составленного плана фармаконадзора, предусматривающего активное пострегистрационное отслеживание безопасности их применения в реальных условиях. Технически сложно и экономически затратно воссоздавать биопрепараты, поскольку сложные белки синтезируются живыми организмами, подвергшимися генетической модификации. В противоположность этому низкомолекулярные лекарства, производимые из химически синтезированного соединения, можно легко повторить и воспроизвести со значительно более низкими затратами. Чтобы биоаналоги дошли до пациентов, необходимо, опираясь на комплекс данных о клинических, доклинических, функциональных аналитических и конформационных характеристиках, доказать, что они максимально идентичны первоначальному инновационному биологическому препарату.[16][17]
Как правило, после того как FDA выпустила лекарство на рынок, его безопасность и эффективность подлежат переоценке каждые шесть месяцев в первые два года продажи. Впоследствии переоценка проводится на ежегодной основе, а результаты оценки подлежат доведению до регулирующих инстанций, таких как FDA. В отличие от генериков, фармаконадзор за биоаналогами по степени своей строгости соответствует требованиям, предъявляемым к фармаконадзору за референтным препаратом (это существенно отличает фармаконадзор за биоаналогами от фармаконадзора за генериками, который достаточно прост и в целом ориентируется на ситуацию с пострегистрационной безопасностью оригинального низкомолекулярного лекарства). Так, заявление о регистрации биоаналогов, одобряемых в Евросоюзе EMA по централизованной процедуре, должно содержать план управления рисками (ПУР), а держатели разрешений на их продажу обязаны предоставлять регулярные обновляемые отчеты о безопасности после выхода препарата на рынок.[18] ПУР содержит характеристику профиля безопасности лекарства, меры по минимизации рисков его применения, план предлагаемых фармаконадзорных исследований, а также меры оценки эффективности принимаемых мер по минимизации рисков.
Проведен ряд ФК-исследований, например исследований, организованных Комитетом по лекарственным препаратам для медицинского применения (КМЛП) EMA, при различных состояниях: антитела оригинального препарата в сравнении с антителами биоаналога, комбинированная терапия в сравнении с монотерапией, различные заболевания и т. д. в целях верификации фармакокинетической сопоставимости биоаналога по отношению к референтному лекарственному препарату на достаточно чувствительной и однородной популяции. Примечательно, что если можно доказать сопоставимость биоаналога и референтного препарата по структуре и функциям, фармакокинетическим профилям и фармакодинамическим эффектам и (или) по эффективности, можно также ожидать, что нежелательные лекарственные, обусловленные избыточными фармакологическими эффектами, будут иметь схожую частоту возникновения.
Одобренные биоаналоги в Евросоюзе
По состоянию на май 2019 года Европейская комиссия по результатам экспертизы EMA одобрило 54 биоаналога. Одобрены биоаналоги следующих действующих веществ: адалимумаб, бевацизумаб, инфликсимаб, ритуксимаб, трастузумаб, этанерцепт; эпоэтины альфа и зета; филграстим и пэгфилграстим; эноксапарин натрия; терипаратид; фоллитропин альфа; инсулин лизпро и инсулин гларгин; соматропин.[19]
Соединенные Штаты Америки
Закон о ЦКИБ
Закон о ценовой конкуренции и инновациях биопрепаратов от 2009 г. (Закон и ЦКИБ) был первоначально проспонсирован и внесен на рассмотрение 26 июня 2007 г. сенатором Тедом Кеннеди (демократ от Массачусетса). Официально закон принят в составе Закона о защите пациентов и доступной помощи, подписанного президентом Бараком Обамой 23 марта 2010 г. Закон о ЦКИБ внес изменения в Закон о службе здравоохранения (Закон о СЗО), создав сокращенный путь одобрения биологических препаратов, подтвердивших свой высокую аналогичность (биоаналогичность) по отношению к одобренному Администрацией по продуктам питания и лекарствам (FDA) биологическому препарату. Концептуально Закон о ЦКИБ схож с Законом о ценовой конкуренции и восстановлении сроков действия патентов на лекарства от 1984 г. (также называемым Законом Хэтча-Ваксмана), который создал в составе Федерального закона о продуктах питания, лекарствах и косметике (FD&CA) правовой механизм одобрения воспроизведённых лекарств. Закон о ЦКИБ согласуется с имеющей длительную историю практикой FDA, позволяющую разработчику и регулятору опираться на уже известные о лекарстве сведения, тем самым экономя время и ресурсы и избегая ненужного дублирования испытаний на людях и животных. По состоянию на май 2019 FDA в общей сложности выпустила 8 методических документов, пять из которых приняты в окончательной редакции, а три — в виде проектов.[20]
В 2018 г. FDA разработала План работ по биоаналогам для исполнения положений Закона о ЦКИБ, включая ограничение злоупотребления системой Стратегий оценки и ослабления рисков (REMS; американский термин для обозначения фармаконадзора) в целях «озеленения», а также переходу к регулированию препаратов инсулина и гормона роста человека в качестве биопрепаратов, нежели лекарств.[21]
Исключительность данных
Исключительность экспериментально полученных данных является важным элементом поправки, внесенной Законом о защите пациентов и доступной помощи в отношении биоаналогов. Это срок между одобрением FDA оригинального препарата и сроком подачи сокращённого досье на биоаналог, опирающегося на данные оригинального разработчика. Исключительность данных направлена на защиту инноваций и на признание длительного, дорогостоящего и рискованного процесса разработки, требуемого для получения разрешения FDA на вывод лекарства на рынок. Срок исключительности данных критичен для будущих биопрепаратов. Некоторые предложения касательно исключительности данных в последних законодательных инициативах доходили до 14 лет, однако Закон о защите пациентов и доступной помощи в итоге предоставляет 12-летнюю защиту с момента одобрения FDA.[22] Этот срок предназначен для компенсации потенциальных недостатков патентной защиты биопрепаратов. Исключительность данных исчисляется со дня одобрения препарата, при этом срок охраны течет параллельно с любым остаточным сроком патентной охраны биопрепарата. Таким образом, исключительность данных предоставляет инновационному разработчику дополнительную охрану, если остаточный срок действия патента короче, чем срок исключительности данных в момент одобрения (что может иметь место вследствие длительных доклинических и клинических изысканий, требуемых для получения одобрения FDA), или если условия патента были преодолены биоаналогом еще до истечения срока действия патента.
Номенклатура
Всемирная организация здравоохранения (ВОЗ) и FDA несколько лет вели работу над присвоением некоммерческих названий биоаналогам. В январе 2017 г. FDA опубликовала указания для отрасли по данному вопросу.[23] Если резюмировать, руководство предусматривает присвоение четырехбуквенного суффикса некоммерческому названию оригинального препарата, чтобы различать инновационные лекарства между собой и отличать их от соответствующих биоаналогов. Вместе с тем ВОЗ, мотивируя свое решение нежеланием препятствовать конкуренции, отказалась от включения в некоммерческие названия биопрепаратов уникальных модификаторов, которые позволяли бы различать биопрепараты разных производителей. Таким образом, система присвоения МНН в отношении биопрепаратов не отличается от таковой, действующей в отношении лекарств на основе низкомолекулярных действующих веществ.
Биоаналоги, одобренные в США[24]
| Дата одобрения FDA | Биоаналог | Оригинальный препарат |
|---|---|---|
| март 2015 г. | филграстим-sndz/Zarxio | филграстим/Neupogen |
| апрель 2016 г. | инфликсимаб-dyyb/Inflectra | инфликсимаб/Remicade |
| август 2016 г. | этанерцепт-szzs/Erelzi | этанерцепт/Enbrel |
| сентябрь 2016 г. | адалимумаб-atto/Amjevita | адалимумаб/Humira |
| май 2017 г. | инфликсимаб-abda/Renflexis | инфликсимаб/Remicade |
| август 2017 г. | адалимумаб-adbm/Cyltezo | адалимумаб/Humira |
| сентябрь 2017 г. | бевацизумаб-awwb/Mvasi | бевацизумаб/Avastin |
| декабрь 2017 г. | трастузумаб-dkst/Ogivri | трастузумаб/Herceptin |
| декабрь 2017 г. | инфликсимаб-qbtx/Ixifi | инфликсимаб/Remicade |
| май 2018 г. | эпоэтин альфа-epbx/Retacrit | эпоэтин альфа/Procrit |
| июнь 2018 г. | пэгфилграстим-jmdb/Fulphila | пэгфилграстим/Neulasta |
| июль 2018 г. | филграстим-aafi/Nivestym | филграстим/Neupogen |
| октябрь 2018 г. | адалимумаб-adaz/Hyrimoz | адалимумаб/Humira |
| ноябрь 2018 г. | пэгфилграстим-cbqv/ Udenyca | пэгфилграстим/Neulasta |
| ноябрь 2018 г. | ритуксимаб-abbs/Truxima | ритуксимаб/Rituxan |
| декабрь 2018 г. | трастузумаб-pkrb/Herzuma | трастузумаб/Herceptin |
| январь 2019 г. | трастузумаб-dttb/Ontruzant | трастузумаб/Herceptin |
| март 2019 г. | трастузумаб-qyyp/Trazimera | трастузумаб/Herceptin |
| апрель 2019 г. | этанерцепт-ykro/Eticovo | этанерцепт/Enbrel |
| июнь 2019 г. | трастузумаб-anns/Kanjinti | трастузумаб/Herceptin |
По аналогии с Оранжевой книгой (официально называемой «Одобренные лекарственные препараты с оценками их терапевтической эквивалентности»), содержащей перечень зарегистрированных FDA лекарственных препаратов с оценкой их терапевтической эквивалентности/взаимозаменяемости и издаваемой ведомством ежегодно с 1980 г., начиная с 2015 г. FDA ведет так называемую Пурпурную книгу.[25] Пурпурная книга, официально называемая «Перечни лицензированных биологических препаратов с исключительностью референтных препаратов и оценками биоаналогичности или взаимозаменяемости», представляет собой два перечня, ведомых двумя центрами FDA, отвечающими за регулирование биопрепаратов: Центром экспертизы и изучения лекарств (CDER) и Центром экспертизы и изучения биопрепаратов (CDER).[26] Перечень Центра экспертизы и изучения лекарств содержит главным образом охарактеризованные терапевтические белковые препараты, входящие в его юрисдикцию.[27] Перечень Центра экспертизы и изучения биопрепаратов содержит вакцины, анатоксины, аллергены, белки плазмы (например, альбумин, факторы свертывания крови, антитромбин III, ингибитор C1-эстеразы, фибриновый клей, иммуноглобулины), гетерологичные иммуноглобулины и иммуносыворотки, генотерапевтические препараты и препараты клеточной терапии и некоторые другие продукты, регулируемые им.[28]
Взаимозаменяемость
Наиболее понятное и простое определение взаимозаменяемости биоаналогов содержится в американском Законе о службе здравоохранения, секция 351(i)(3) которого устанавливает, что взаимозаменяемость имеет место, когда "биологическим препаратом можно заменить референтный препарат без вмешательства медицинского работника, назначившего референтный препарат". При этом секция 351(k)(4) Закона содержит научно-регуляторные стандарты взаимозаменяемости и закрепляет, что биоаналогичный препарат взаимозаменяем с референтным биологическим препаратом, если можно ожидать, что биоаналог будет приводить к такому же клиническому результату, что и референтный препарат у любого рассматриваемого пациента и что в случае биологического препарата, вводимого индивиду неоднократно, риск с точки зрения безопасности или сниженной эффективности в связи с чередованием применения биоаналога и референтного препарата либо переключением с одного на другой не выше, чем риск применения референтного препарата без такого чередования или переключения.[29] Таким образом, правила США предусматривают сначала подтверждение биоаналогичности, а только потом — с помощью отдельного комплекса исследований — взаимозаменяемости биоаналога с референтным препаратом.
Администрация по продуктам питания и лекарствам (FDA) выработала правила подтверждения взаимозаменяемости биоаналога с соответствующим ему референтным биологическим препаратом, предусматривая проведение исследования (или нескольких исследований) переключения. Предлагаемый FDA дизайн представляет собой исследование со вступительным периодом вмешательства референтным препаратом с последующим рандомизированным периодом в двух группах, одна из групп которого предусматривает переключение между предлагаемым взаимозаменяемым препаратом и референтным препаратом (группа переключения), а другая остается в качестве группы непереключения, получающей только референтный препарат (группа непереключения).[30]
Европейская сеть регулирования лекарств (EMRN)[31], куда входят Европейское агентство по лекарствам (EMA), национальные уполномоченные органы государств — членов Евросоюза, соответствующее подразделение Еврокомиссии, EDQM и т. д., взаимозаменяемость биоаналогов с точки зрения чередования применения референтного препарата и биоаналога не определяет. В базовом руководстве EMA по биоаналогам «Аналогичные биологические лекарственные препараты» указано: «Экспертиза биоаналогичных лекарств в регистрационных целях, проводимая EMA, не предусматривает предоставления рекомендаций относительно взаимозаменяемости биоаналога с его референтным лекарством. Политика замещения находится в ведении государств — членов ЕС».[14] Признавая биоаналогичность EMA подспудно подразумевает и взаимозаменяемость, однако детально не останавливается на вопросах чередования и переключения. Государства — члены ЕС не имеют собственных научных руководств, которые содержали бы подходы к определению взаимозаменяемости с точки зрения переключения или чередования.
Большое внимание в Европейском союзе уделяется пострегистрационному наблюдению за биоаналогами. FDA в своем документе как бы косвенно критикует этот подход, указывая, что «мы, как правило, не ждём, что пострегистрационные данные позволят получить достаточные сведения, касающиеся влияния на клиническую фармакокинетику (ФК) и фармакодинамику (ФД) при переключении или чередовании применения предлагаемого взаимозаменяемого препарата и референтного препарата, которые, по нашему мнению, являются важными учитываемыми исследуемыми конечными точками в исследованиях переключения…», но при этом «в определенных ситуациях данные пострегистрационного наблюдения о лицензированном биоаналогичном препарате в дополнение к должным образом спланированному исследованию переключения могут потребоваться для снятия неопределенности относительно подтверждения взаимозаменяемости и пополнения совокупности доказательств для обоснования подтверждения взаимозаменяемости».[30]
Рыночные вопросы
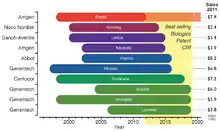
Законодательные требования к выводу на рынок вместе с дорогостоящими процессами производства толкают вверх расходы на разработку биоаналогов: затраты на одну молекулу могут доходить до 75–250 млн. долл. США.[32] Такой барьер для входа на рынок влияет не только на желание компаний производить их, но также может препятствовать доступности недорогих альтернатив для учреждений здравоохранения, субсидирующих лечение своих пациентов. Даже несмотря на рост рынка биоаналогов, снижение цен на биологические лекарства с истекающими патентами будет не столь значительной, как для других воспроизведенных лекарств; по оценочным расчетам цена на биоаналогичные препараты будет лишь на 15–35 % ниже, чем на соответствующие оригинальные препараты.[32] Биоаналоги привлекают внимание рынка в связи с надвигающимся патентным обрывом, который подвергнет риску около 36 % 140-миллиардного (в долл. США) рынка биологических лекарств (по состоянию на 2011 г.); это только если рассматривать 10 самых продаваемых препаратов.[32]
Глобальный рынок биоаналогов в 2013 г. составил 1,3 млрд. долл. США с ожидаемым ростом до 35 млрд. долл. США в 2020 г., который будет обеспечен истечением действия патентов на еще 10 биопрепаратов-блокбастеров.[33]
См. также
Ссылки
- Blanchard, A., Helene D'Iorio and Robert Ford. "What you need to know to succeed: Key trends in Canada's biotech industry " Insights, spring 2010
- Nick, C. The US Biosimilars Act: Challenges Facing Regulatory Approval (англ.) // Pharm Med : journal. — 2012. — Vol. 26, no. 3. — P. 145—152. — doi:10.1007/bf03262388. Архивировано 16 января 2013 года.
- Lamanna, William C.; Holzmann, Johann; Cohen, Hillel P.; Guo, Xinghua; Schweigler, Monika; Stangler, Thomas; Seidl, Andreas; Schiestl, Martin. Maintaining consistent quality and clinical performance of biopharmaceuticals (англ.) // Expert Opinion on Biological Therapy : journal. — 2018. — 10 January (vol. 18, no. 4). — P. 369—379. — ISSN 1744-7682. — doi:10.1080/14712598.2018.1421169. — PMID 29285958.
- Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. www.fda.gov.
- Biosimilar Approval Status in the US: FDA Filing Dates and Actions. Biosimilars Review & Report (26 Arpil 2019).
- EMEA Guideline on Similar Biological Medicinal Products, CHMP/437/04 London, 30 October 2005.
- US Senate Committee on the Judiciary, Testimony of Dr. Lester Crawford, Acting Commissioner, FDA June 23, 2004 (недоступная ссылка). Дата обращения: 19 мая 2019. Архивировано 28 декабря 2016 года.
- Hearing: Assessing the Impact of a Safe and Equitable Biosimilar Policy in the United States. Subcommittee on Health Wednesday, May 2, 2007 Архивировано 22 сентября 2007 года.
- FDA page on "Follow-On Protein Products: Regulatory and Scientific Issues Related to Developing".
- FDA page on "Approval Pathway for Biosimilar and Interchangeable Biological Products Public Meeting".
- FDA Response to three Citizen Petitions against biosimilars.
- FDA page on "FDA approves first biosimilar product Zarxio".
- Martina Weise. Biosimilars: the science of extrapolation (англ.) // Blood. — American Society of Hematology, 2014. — 8 October (vol. 124, no. 22). — P. 3191—3196. — doi:10.1182/blood-2014-06-583617. — PMID 25298038. (недоступная ссылка)
- EMA guideline on similar biological medicinal products. Дата обращения: 19 мая 2019. Архивировано 5 июля 2019 года.
- Warren, J. B. Generics, chemisimilars and biosimilars: is clinical testing fit for purpose? (англ.) // Br J Clin Pharmacol : journal. — 2013. — Vol. 75, no. 1. — P. 7—14. — doi:10.1111/j.1365-2125.2012.04323.x. — PMID 22574725.
- Wang, X. Higher-Order Structure Comparability: Case Studies of Biosimilar Monoclonal Antibodies (англ.) // BioProcess International : journal. — 2014. — 1 June (vol. 12, no. 6). — P. 32—37.
- Declerck P. J. Biosimilar monoclonal antibodies: a science-based regulatory challenge (англ.) // Expert Opin Biol Ther : journal. — 2013. — February (vol. 13, no. 2). — P. 153—156. — doi:10.1517/14712598.2012.758710. — PMID 23286777.
- EMA guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Дата обращения: 19 мая 2019. Архивировано 5 июля 2019 года.
- Biosimilars approved in the EU. European Medicines Agency. Дата обращения: 19 мая 2019.
- All Guidances for Drugs; search for ‘biosimilars’.
- Commissioner, Office of the Press Announcements - Statement from FDA Commissioner Scott Gottlieb, M.D., on new actions advancing the agency's biosimilars policy framework (англ.). www.fda.gov. Дата обращения: 19 мая 2019.
- 42 U.S. Code § 262 - Regulation of biological products. LII / Legal Information Institute.
- U.S. Food and Drug Administration | Nonproprietary Naming of Biological Products; Guidance for Industry (17 мая 2019). Дата обращения: 17 мая 2019.
- Biosimilar Product Information - FDA-Approved Biosimilar Products. www.fda.gov.
- Background Information: Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations (Purple Book). www.fda.gov.
- Purple Book: Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations. www.fda.gov.
- CDER List of Licensed Biological Products: List of Licensed Biological Products with (1) Reference Product Exclusivity and (2) Biosimilarity or Interchangeability Evaluations to Date. www.fda.gov.
- CBER List of Licensed Biological Products: List of Licensed Biological Products with (1) Reference Product Exclusivity and (2) Biosimilarity or Interchangeability Evaluations to Date. www.fda.gov.
- 42 USC Chapter 6A, Subchapter II: General Powers and Duties, Part F—Licensing of Biological Products and Clinical Laboratories, subpart 1—biological products, §262. Regulation of biological products (42 U.S.C §262(k)(4)). www.fda.gov.
- Considerations in Demonstrating Interchangeability With a Reference Product Guidance for Industry. www.fda.gov.
- European medicines regulatory network. www.ema.europa.eu.
- Calo-Fernández B., Martínez-Hurtado J. Biosimilars: Company Strategies to Capture Value from the Biologics Market (англ.) // Pharmaceuticals : journal. — 2012. — December (vol. 5, no. 12). — P. 1393—1408. — doi:10.3390/ph5121393. — PMID 24281342.
- Biosimilars and Follow-on-Biologics Market to Hit $35 Billion Globally by 2020. Pharmaceutical Technology (28 августа 2015).
Дополнительные материалы
- Ниязов, Равиль Р.; Рождественский, Дмитрий А.; Васильев, Андрей Н.; Гавришина, Елена В.; Драницына, Маргарита А.; Куличев, Дмитрий А. Регуляторные аспекты регистрации воспроизведенных и гибридных лекарственных препаратов в Евразийском экономическом coюзе // Журнал «Ремедиум» : журнал. — 2018. — Сентябрь (т. 17, № 7—8). — С. 6—19. — doi:10.21518/1561-5936-2018-7-8-6-19. — PMID 26678619.

- Биологические препараты. Pharmaceutical Education and Development (PhED) (3 октября 2016). Дата обращения: 18 мая 2019.
- Udpa, Natasha; Million, Ryan P. Monoclonal antibody biosimilars (англ.) // Nature Reviews Drug Discovery : journal. — 2015. — 18 December (vol. 15, no. 1). — P. 13—4. — doi:10.1038/nrd.2015.12. — PMID 26678619.
- Jelkmann, Wolfgang. Biosimilar epoetins and other "follow-on" biologics: update on the European experiences (англ.) // American Journal of Hematology : journal. — 2010. — Vol. 85, no. 10. — P. 771—780. — doi:10.1002/ajh.21805. — PMID 20706990.
- New guide on biosimilar medicines for healthcare professionals (англ.) // Prepared Jointly by the European Medicines Agency and the European Commission : journal.
- "Правила проведения исследований биологических лекарственных средств Евразийского экономического союза". Утверждены решением Совета Евразийской экономической комиссии от 3 ноября 2016 года № 89